HPCチャレンジを動かして、大まかなARMのcpuの性格が分ったという事で、では、実アプリじゃどうなのか、という話になりました。
OSは共通、さらに、gccやclang、flangが使用可能という事で、ソースがあるlinux上の大抵のアプリはポート出来るとは思いはするものの、やはり、ポーティングされた例が多いものである方が成功する可能性は高いと考えられます。ポーティング例が多く、ソースが付属しているとなると、やはりOpenSouce系のアプリが良いでしょう。という訳で、gromacsをビルドして動かしてみる事にしました。
gromacsを選択する理由として
1、OpenSourceであり、ユーザー数も多く、様々なアーキテクトで動作実績がある。
2、gromacsはMDエンジン部分に、CPU依存コードが使用可能ですが、ARM64のコードがあるので、性能もそこそこ期待出来る。
3、blasやFFTに依存する部分が少ない。
という3つの理由から選択しました。
まあ、上記の理由も今となれば、という感じもあるのですが。
というのも、ver3.0くらいまでの頃は、CPMDがソースが公開されたのにgromosは公開されず、CPMDとの連携で欲しいという場合がメインであったのと、正直に当時の状況はというと、ビルドシステムの出来が、アレで、そのままではまともにビルド出来ず、MDの知識以外にgnu makeやm4、libtoolなんかの知識が結構、必要であった為、ユーザーが本邦で豊富だったかというと微妙な話でした。弊社でインストールサポートサービスを行なったところ、あれよあれよと引き合いが来て吃驚した記憶があります。
さらに、amber force fieldがamber toolsがOpenSource化した事によって、gromacsで使用可能になった事も追い風になったと思います。もっとも、本邦の場合、液晶の基礎研究で使用されて名が広まったという日本ならではの零れ話もあったりもします。
そういった理由もあるのですが、他に、gromacsの開発が述べた地上最速のMDを作るという目標に誘われて、沢山の趣味のプログラマが開発に参加したという点は、実は大きいかもしれません。ポーティングが豊富なのは、これが最大の理由でしょう。新しいアーキテクトで少々重い計算をさせてみる場合、gromacsがポートされる場合は比較的多いように思います。
もっとも、これは痛し痒しな面もあって、なまじ最速を目指す人が増えたせいか、計算結果はどうでもいいという場合もそこそこ開発者の中にはいらっしゃるみたいで、最近は後方互換性を維持しようとしない傾向が高いように思います。まあ、CPMDと連動させて動作させたいという場合はほぼいないのでしょうけど、後方互換性より、速度、新機能に注力するあまり、過去の公式ベンチマークが動かないといった本末転倒したかのような面はあります。
新機能が増えるのは良いのですが、後方互換性を気にしないで実装しちゃう場合も多く、ver5より前のインプットは、動かすのに修正が必須というのが実際だったりします。そうなると、果してどのくらい性能が上がったかが判断出来ないですよね。
実計算で使用する場合、新規にgromacsを使用するのであればそれで問題無いでしょうけど、継続して使用している場合、verが変る度にマニュアルを精査しなくてはいけないのは、実アプリとしては如何なものか、とは思います。こうしたアプリとしては例外的に情報豊富なマニュアルがありはするのですが、1冊の本規模のマニュアルをバージョンアップの度に首っぴきになるのが嬉しいかどうかは疑問は感じざるえません。まあ、pythonほどでは無いとは思いますけどね。pythonのあのマイナーバージョンなどでの互換性の無さは、開発者を締め殺すに十分な凶暴性ですよ。
blasやFFTに依存しないのは、開発環境がgcc + gnu makeだけでも可能なように、という面はあるのかもしれませんが、これも最速を目指す過程で、通常ならblasを使うとこまで手を入れているみたいです。その為、外部の高性能のblasやFFTを使用しても、ほぼ効果が無いのですが、逆にblasやFFTを高速化したりしないでgromacsに注力すれば良いという事で、ポーティングは容易かどうかは別としてポートされる場合は増えたと思います。重いところは有料のblasで高速化してというのは定番ですが、新しいアーキテキトの場合、それが出来ない場合も多いので、こうした試験には向いているところですね。
さて、ビルドですが、gromacsは計算エンジンのコア部分がCPU依存コードが使用可能で、その為、AVXとSSEですらコードが異なる為、注意が必要です。SIMD対応はARMは、ARM_NEONとARM_NEON_ASIMDの2種類です。残念ながら、ここで使用したgromacsのバージョンではthunderX2の128bit simdは使用出来ないみたいです。neonは32bitのもので、double floatがありませんから、使用可能なのは、ASIMDの方となります。恐らくARMv8-aのSIMDという事なんでしょう。
実は、nb_kernelの部分でもintelのCPUは固有のSIMDに依存したコードがあるのですが、残念ながら、このバージョンのgromacsではARMには未対応のようで、汎用コードを使用せざるえません。とりあえず、ビルドして使用出来るというレベルですね。gromacsはこうした部分のチューニングが盛んなので、マイナーバージョンでの違いでも、最新のものなら対応しているかもしれません。もっとも、最新の物は、経験的に大変、不安定で、マイナーナンバーが3以降くらいにならないとビルドする気になれなかったりします。
使うSIMDの選択とMPIをかっちり作ってあれば、gromacsのclangを使用したビルドはそう難しくはありませんでした。gromacsに慣れていないと意図しない形にビルドされたりもするのですが、そこは慣れで対応です。というのは、cmakeのビルドスクリプトに穴が多いんで、結構、gromacs特有の経験が必要だったりします。
SIMD依存コードがARMv8のneon64のものとnb_kernelが汎用コードなので、intel CPUほど速度は期待出来ない状態ではあるのですが、動作に問題はありませんでした。
もっとも、そう期待は出来ないとはいうものの、gromacsは速度重視なのでオプティマイズをしっかりやっておく事も大事です。オプティマイズをしっかりやらないと、大変な低速に終ってしまいますからね。
どのくらい大変になってしまうかが次の図です。
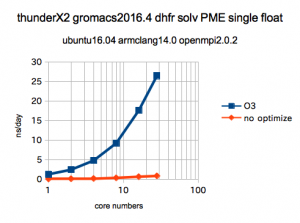
armclangの性能が結構、良いというのもありますが、このようにオプティマイズが効果があるコードである事が分ります。
このベンチはジヒドロ葉酸レクターゼをPMEで計算したもので、以前、gromacs-GPU用の公式ベンチとして公開されていたものです。
http://www.gromacs.org/Documentation/Installation_Instructions_4.5/GROMACS-OpenMM#GPU_Benchmarks
このような蛋白質が実際の計算では以下の図のように水溶液中にあります。

実際の研究に即したベンチだと考えられるので、参考になりますよね。
