
こんにちは!HPC事業部の技術グループのnabeoです。
半経験的タイトバインディングのソフトウェア xTB は、軽量な量子化学計算エンジンとして様々な論文で見かけるようになりました。そこで、GRRMがxTBを使うように連携させて反応経路探索をやってみたところ、一晩で1万個を超える分子構造を探索することができました。以下では、その構築/実行方法と結果を書いています。
環境構築
xTB のインストール
$ wget https://github.com/grimme-lab/xtb/releases/download/v6.7.1/xtb-6.7.1-linux-x86_64.tar.xz $ tar xf xtb-6.7.1-linux-x86_64.tar.xz $ mv xtb-dist xtb-6.7.1 $ vim ~/.bashrc export PATH="${HOME}/xtb-6.7.1/bin:${PATH}" export LD_LIBRARY_PATH="${HOME}/xtb-6.7.1/lib:${LD_LIBRARY_PATH}" export OMP_NUM_THREADS=1,1
ちなみに xtb はOpenMP並列プログラムなので環境変数 OMP_NUM_THREADS でスレッド並列数を指定することが可能ですが、200原子くらいまでの軽い計算なら一瞬で終わるので、並列オーバーヘッドを避けるようにスレッド並列数は 1 にしました。
grrm2xtb のインストール
続いて xTB と GRRM をつなぐインタフェースプログラムをインストールします。今回、このインタフェースには 、GitHub で公開されている、第三者作成の grrm2xtb を使わせてもらいました。作成者の npe1011 さん、ありがとうございます。
このプログラムはC++で書かれていて軽量に動作するのが特徴ですが、一部がC++17を使って書かれているために、コンパイルするためにはC++17対応のコンパイラが必要です。そこで今回は、コンパイラにインテル oneAPI 2025.2 の icpx を使用しました(ダウンロードしてインストーラに沿って入れるだけなので、oneAPI 2025.2 のインストール手順は割愛します)。
grrm2xtb は icpx にPATHが通っている状態でmakeするだけですぐにコンパイルできますが、static link用のライブラリが今回の計算機に足りなかったので、static link させるためのコンパイラオプション -static を外して、dynamic link にしてビルドしました。実行計算機上でのコンパイルなので、dynamic link でも問題ありません。
$ git clone https://github.com/npe1011/grrm2xtb_cpp.git $ vim makefile CXXFLAGS = -std=c++17 -O3 -I./src #CXXFLAGS = -std=c++17 -O3 -static -I./src $ make $ vim ~/.bashrc export PATH="${HOME}/grrm2xtb_cpp:${PATH}"
計算実行
今回は例としてウェーラー合成のSC-AFIR探索計算をRCMC付きで実行してみました。
GRRM 用インプットファイル作成(wohler_scafir_rcmc.com)
%link=non-supported を加え、Options より下には sublink=grrm2xtb を記述するのがポイントです。こうすることで、GRRM23 が量子化学計算エンジンとして grrm2xtb を呼び出すようになります(そして grrm2xtb が xTB を呼び出して Energy・Gradient・Hessian を実行します)。%link=non-supported # SC-AFIR/wB97XD/Def2SVPP scrf=(smd,solvent=water) 0 1 N 0.07627118 -2.83050843 0.00000000 1 H 0.40959278 -3.77332162 0.00000000 1 H 0.40961039 -2.35910806 0.81649658 1 H 0.40961039 -2.35910806 -0.81649658 1 H -0.92372882 -2.83049598 0.00000000 1 O 1.83235868 -3.55638704 0.51820322 2 C 3.26235868 -3.55638704 0.51820322 2 N 4.40895868 -3.55638704 0.51820322 2 H 5.09666726 0.18549783 3.27098966 3 H 3.81621267 1.09043366 3.27098966 3 O 4.13666726 0.18549783 3.27098966 3 H 4.68144098 -6.12407202 -1.61170341 4 H 3.40098639 -5.21913619 -1.61170341 4 O 3.72144098 -6.12407202 -1.61170341 4 Options NRUN=256 GauProc=16 GauMem=300 Add Interaction GAMMA=200.0 END Clustering=Connectivity MatchDecTarget=1-14 MatchDecScale=3.0 8.0 NOFC EigenCheck Derivative=Force MinFreqValue=50.0 TrafficVolCheck TimeScale=3600.0 SIML_Temperature=300.0 350.0 400.0 SIML_pLatest=420 SIML_tLargest=140 rTemperature=4000.0 DownDC=15 sublink=grrm2xtb
grrm2xtb の環境変数指定
The calculation settings for XTB (charge, spin multiplicity, solvation, XTB version) need to be given as environmental variables (see below), NOT in the GRRM input. The charge and multiplicity in the GRRM input are neglected and have no effect. In case no environmental variables are set, the default settings are used (charge = 0, multiplicity = 1, gas phase, GFN2). XTB_MULTI should be a spin multiplicity like in Gaussian or GRRM, not Na-Nb (–uhf for XTB). Internally, –uhf is set to multiplicity-1. XTB calculations are done in sub-directories in XTB_SCRATCH_DIR. If not provided, sub-directories are prepared in the GRRM working directory.export XTB_CHARGE=0
export XTB_MULTI=1
export XTB_SOLVATION=ALPB
export XTB_SOLVENT=CH2Cl2
export XTB_PARAM=2
export XTB_SCRATCH_DIR=/path/to/scratch/directory
XTB_PARAM って何だ!?と初見では戸惑ってしまいましたが、ソースコードを確認したところ、これは xtb コマンドの --gfnオプション(GFN-xTBのパラメータ化を指定するオプション)に渡す値でした。そしてデフォルトは2なので、今回はデフォルトのまま(=未指定)にしました。export XTB_CHARGE=0 export XTB_MULTI=1 export XTB_SOLVATION="ALPB" export XTB_SOLVENT="water"
GRRM23の実行
以上が出来たら、GRRM23の計算を投入します。-pのプロセス並列オプションを使って、32並列探索させてみました。これにより網羅的探索において最大32本の反応経路を同時に並列に探索してくれます。
GRRM23p wohler_scafir_rcmc -p32 &
今回の計算は約31時間で完了しました(初期構造をNRUNでランダム生成しているので、同じインプットでも計算時間には多少のばらつきがあると思います)。探索された構造は EQが 3,003個、TSが 1,967個、PTが 20,443個 でした。軽量な分、そう長くない時間でも膨大な数の構造が見つかりますね!
可視化
この探索結果を弊社のGRRMmapで可視化してみました。GRRMmapは、膨大な数の探索結果を可視化・解析するのにきわめて強力です。使い方はコマンドラインで次のように叩くだけ!
GRRMmap wohler_scafir_rcmc
こうすると可視化処理されたデータがHTMLで出力されるので、それをChrome等のWebブラウザで閲覧します。ちなみにこの可視化処理は100秒で完了しました。
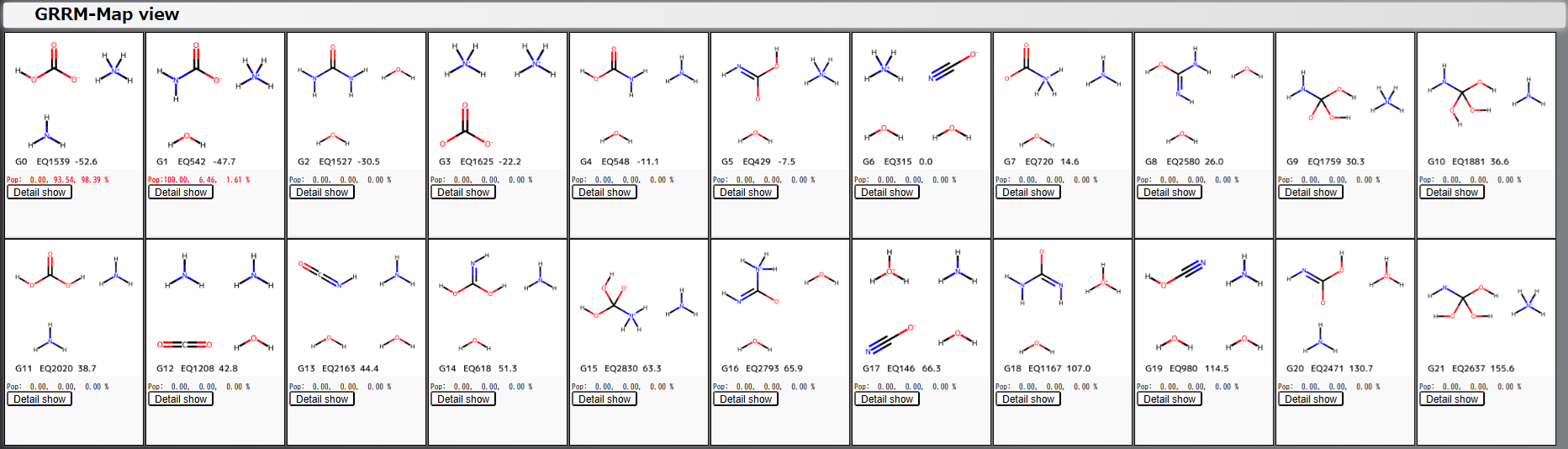
GRRMmapの分子カタログ画面は次のようになりました。GRRMmapでは『同じSMILESになるEQを一つのグループにまとめる』という見せ方をするので、EQ3,003個がグループ22個へと、見るべき情報の量が2桁も圧縮されています。グループごとに、構造式、グループ番号、グループ内でエネルギーが最も低いEQの番号、全EQの中で最低のエネルギー値を0.0としたときの相対エネルギー値[kJ/mol]が表示されています。また、RCMCによって計算されたPopulationの値も赤色の文字で表示されています。
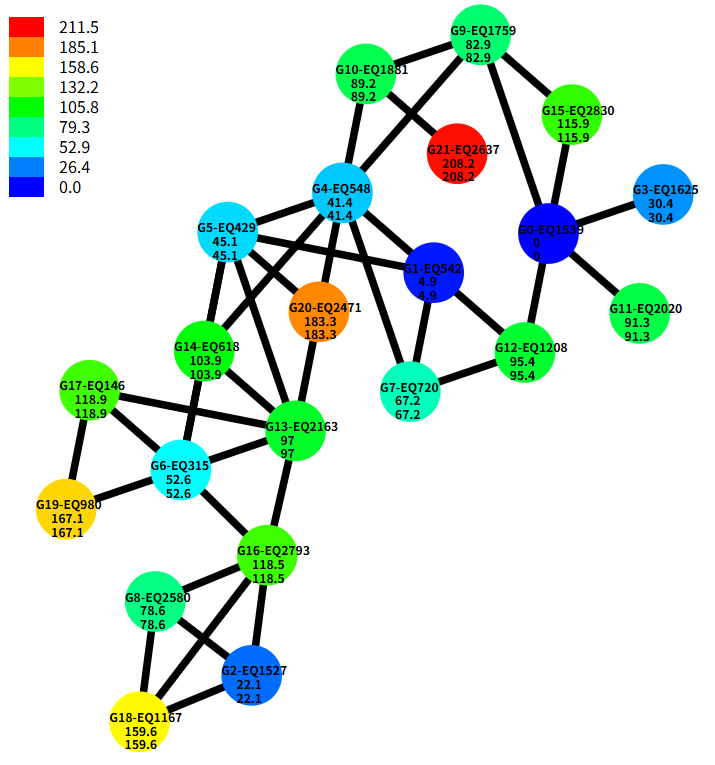
また、反応経路ネットワークを可視化させたところ次の図になりました(●はEQひとつではなく、同一SMILESとなるEQをまとめたグループひとつです)。
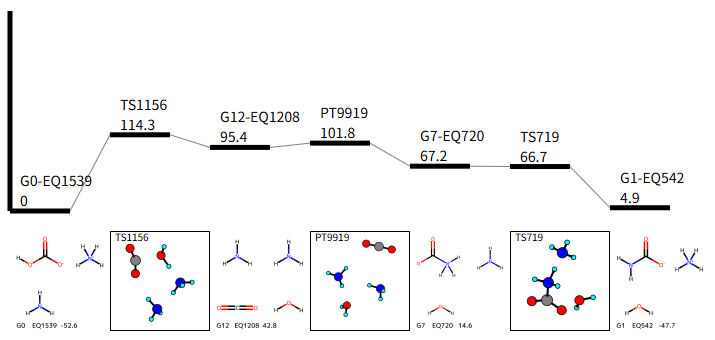
ネットワークグラフの●を2か所クリックすることで、それらの間の最短経路のエネルギー準位図が瞬時に表示されます。G0 と G1 をクリックしたときにRCMCを考慮した経路のエネルギー準位図を以下に示します。間を結ぶTS or PTの構造は Ball-and-stick で3次元表示されており、マウスで回転させて確認することが可能です。
Tips & Take aways
- GRRM with xTB で、もっと大きな分子で探索させたら、xTBの軽量さがさらに際立ちます。そしておそらく、見つかるEQ数・PT数も増えるので、ますますGRRMmapの便利さが感じられるでしょう!
- CPUコア数が少ないPCであっても、xTBで探索させておけば、その結果を高性能計算機へコピーしてGaussianで高い計算レベルで ReStruct 等することで、xTBの探索結果を有効活用できると思います。
Thanks for reading! More HPC in our lives!
