
UMAとは
Meta社のUMA(Universal Model for Atoms)は、Meta社のFAIR(Fundamental AI Research)チームが開発した、分子や材料の化学特性を原子レベルで予測するための汎用AIモデルです。UMA には次の特徴があります。
汎用性:薬剤開発、電池材料、触媒研究など、幅広い分野に対応します。
高速性:従来の分子シミュレーションが数日かかっていたのに対し、UMAでは数秒で完了可能です。
高精度:従来の専用モデルと同等以上の予測精度を持ちます。
UMAは、Metaが公開した大規模な量子化学データセット「Open Molecule 2025 (OMol25)」を含む、過去5年間のデータセットを用いて訓練されています。OMol25には、1億件以上の分子構造データと詳細な化学特性(エネルギー、電荷、スピンなど)が含まれています。このデータセットの構築には、約60億コア時間の計算リソースが投入されました。
Hugging Face で配布されている UMA は、アクセス申請が Hugging Face から許可されると無償で利用することができ、Pythonモジュールの fairchem-core としてインストールして、Pythonの Atomic Simulation Environment (ASE) から呼び出すなどの形態で使用できます。

GRRMと連携させる仕組み
UMA単体をインストールして計算をする手順については こちらのLabCodeさんの記事 や こちらの記事 などが参考になります。ASEのAPI関数である get_potential_energy() や get_forces() を使うことで、UMAで推論された ポテンシャルエネルギー や force を取得できます。こうしたASE APIの活用がUMAとの対話の基本になります。
一方、GRRMには「汎用外部プログラムインターフェース」があります。これは、所定の書式でファイル入出力を行うプログラムを用意しておいて、そのプログラムをGRRMインプットファイルで指定すれば、そのプログラムを(Gaussian等の代わりに)量子化学計算エンジンとして呼び出しながら反応経路探索を行うようにできる仕組みです。そこで、このインターフェース要求仕様に沿うように、GRRMからの計算要求(Energy/Gradient/Hessian)を読み取って、その計算要求を(ASEを介した)UMAで実行し、その計算結果をGRRMが解釈できるように形式を揃えて渡す、という内容のインターフェースプログラムをPythonで自作しました。
これにより、GRRMから「汎用外部プログラムインターフェース」としてこの自作したプログラムを起動させ、それがUMAの推論ルーチンを呼び出す、という連携ができるようになり、Gaussian等の量子化学計算エンジンの代わりにUMAを使ってGRRMの反応経路探索計算を実施できるようになりました。
GRRM ↔ 汎用外部プログラムインタフェース仕様に沿った自作プログラム ↔ UMA
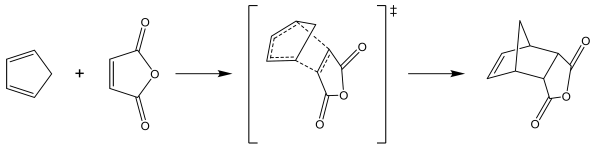
実行結果:Diels-Alder反応
反応経路探索の基本的な例として、Diels-Alder反応の2点間探索を実施してみました。
|
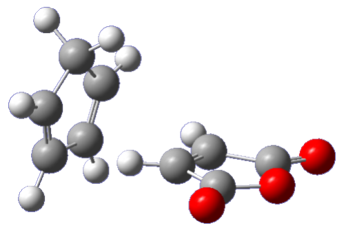
始状態 |
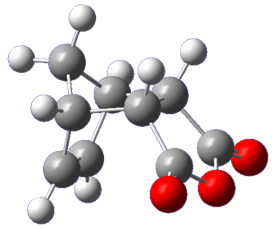
終状態 |
GRRMには GRRM23 を用い、2点間探索として DS-AFIR を用いました。計算レベル(UMAでいうところのtask)には分子系に向いている omol を使用しました。
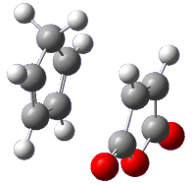
この結果、得られた反応経路(IRC)を次に示します。
| DC | TS | EQ | Animation |
 |
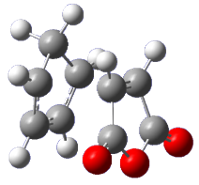 |
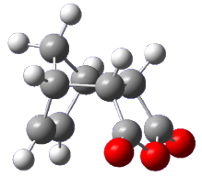 |
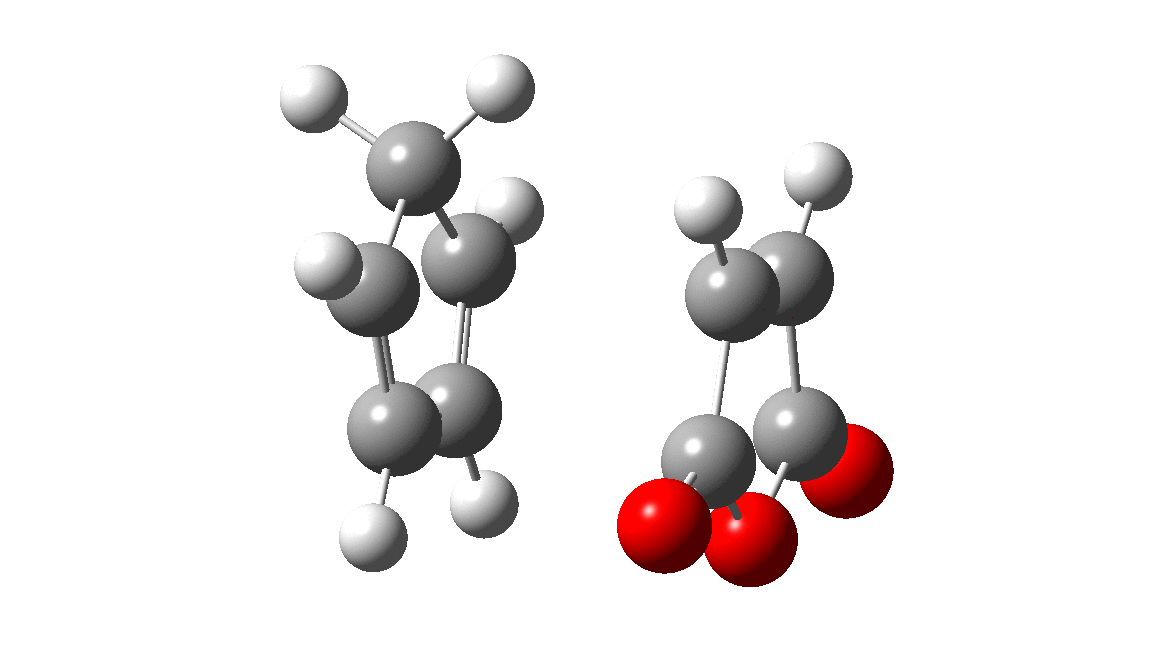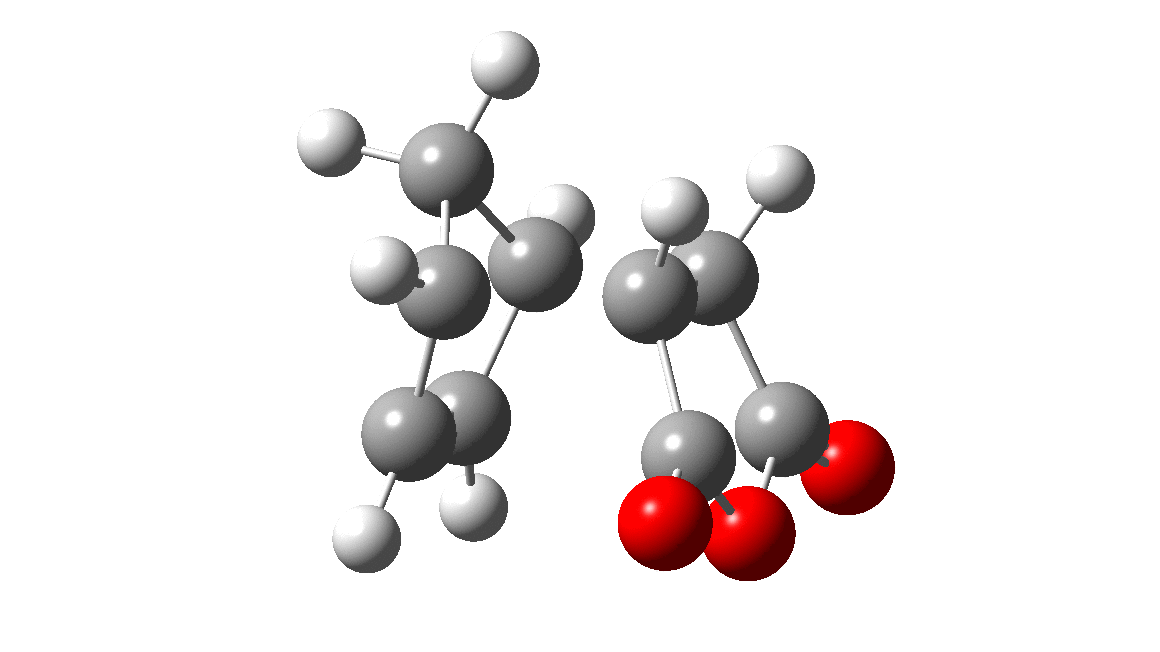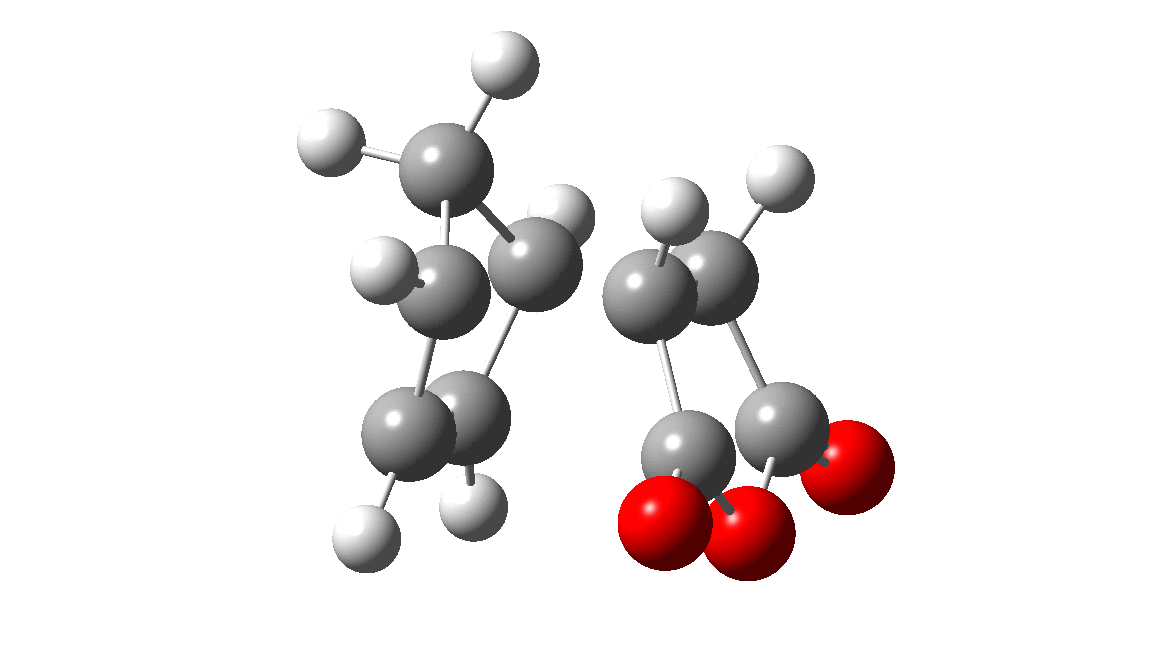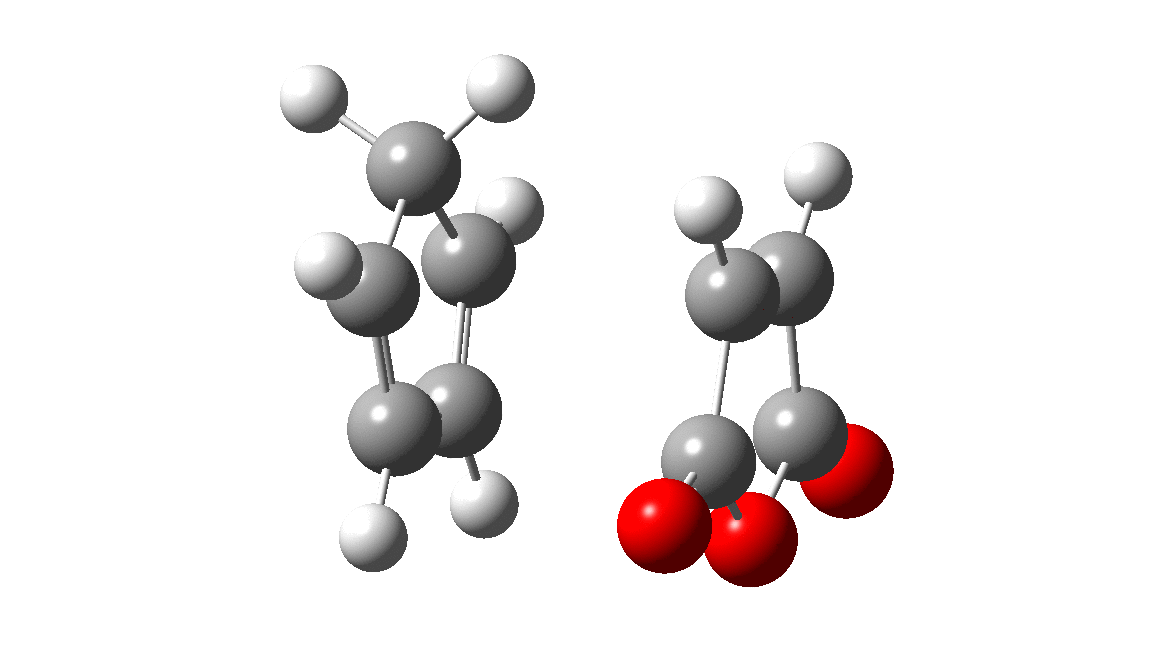 |
既知の反応経路に合致する経路が得られました。
また、IRCに沿ったエネルギープロファイルを次に示します。
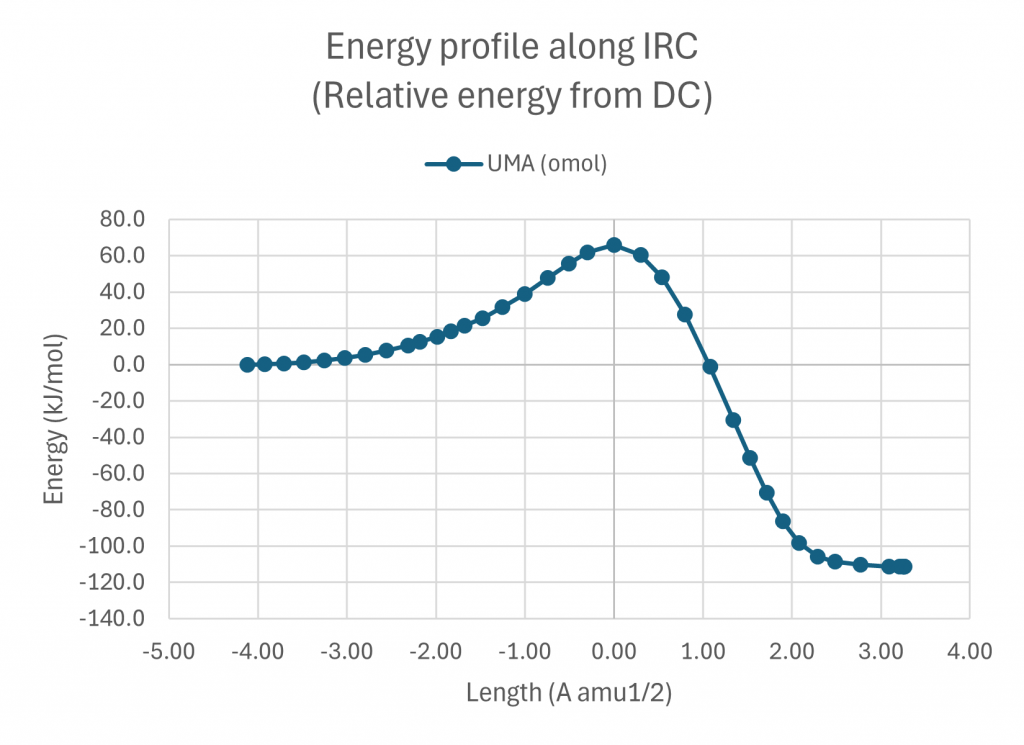
エネルギープロファイルも妥当な形が得られました。
omol の学習データには ORCA 6 実装の ωB97M-V/def2-TZVPD が用いられたそうですが、(詳細はここでは省きますが)その計算レベルに高い精度で一致するエネルギー値がUMAで推論されています。
まとめ
UMAによる機械学習ポテンシャルに基づく推論をGRRMと連成させて、Diels-Alder反応の経路を探索させることができました。機械学習ポテンシャルの強みは計算コストのオーダーの小ささにあり(O(N)に近い)、特に、大きなサイズの分子の高速計算において高い威力を発揮します。大きな分子でのGRRM反応経路探索をぐっと現実的にさせる方法の一つとして、興味深い技術と思います。
