
Science as a Service
Software
計算化学 ソフトウェア
Products
ソフトウェア製品一覧
■ 量子化学
-
- GRRM20 with Matlantishttps://www.hpc.co.jp/chem/software/grrm20-with-matlantis/
-
HPCシステムズ株式会社、Matlantis株式会社およびENEOS株式会社(以下ENEOS)がGRRM20 with Matlantisを共同開発しました...
-
- Gaussianhttps://www.hpc.co.jp/chem/software/gaussian/
-
Gaussianは量子化学計算のデファクト・スタンダードといわれる、米Gaussian社の電子構造モデリングの先端技術を持った量子化学計算プログラムです。実...Gaussian社開発のデファクト・
スタンダード量子化学計算プログラム
-
- GRRMhttps://www.hpc.co.jp/chem/software/grrm-portal/
-
たった1つの分子をインプットするだけで、そこから生成されうるすべての生成物やそれを生成しうるすべての反応物、そしてそれらすべての反応経路を自動的に探索できる...たった1つの分子をインプットするだけで
すべての反応経路を自動探索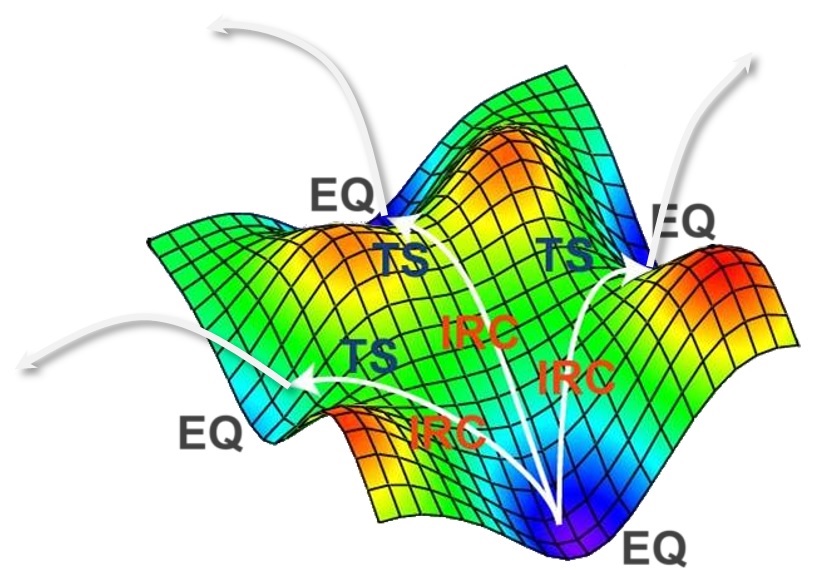
-
- Reaction plus Pro 2 / Express 2https://www.hpc.co.jp/chem/software/react2/
-
これまで反応経路を求めるには経験と勘が必要とされ、特に肝となる遷移状態(TS)構造の最適化には大変な苦労を強いられることが常でした。また、せっかくTSが求ま...ONIOM法を用いた酵素反応等、
Reaction plusではできなかった
様々な反応系の計算が可能に
-
- GRRM26https://www.hpc.co.jp/chem/software/grrm26/
-
たった1つの分子をインプットするだけで、そこから生成されうるすべての生成物やそれを生成しうるすべての反応物、そしてそれらすべての反応経路を自動的に探索できる...たった1つの分子をインプットするだけで
すべての反応経路を自動探索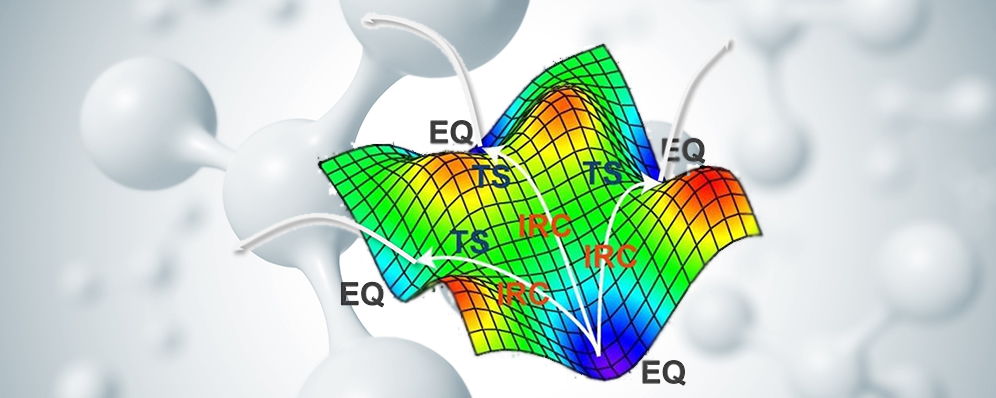
-
- GRRM23https://www.hpc.co.jp/chem/software/grrm23/
-
たった1つの分子をインプットするだけで、そこから生成されうるすべての生成物やそれを生成しうるすべての反応物、そしてそれらすべての反応経路を自動的に探索できる...たった1つの分子をインプットするだけで
すべての反応経路を自動探索
-
- GRRMmaphttps://www.hpc.co.jp/chem/software/grrmmap/
-
GRRMは未知の反応経路を探索する点において大変強力です。一方、計算内容によっては非常に大量の探索結果を生じうるため、探索結果から必要とする情報に短時間で視...GRRMの計算結果を見やすく可視化
大量の探索結果を効率よく閲覧
-
- GRRM20https://www.hpc.co.jp/chem/software/grrm20/
-
たった1つの分子をインプットするだけで、そこから生成されうるすべての生成物やそれを生成しうるすべての反応物、そしてそれらすべての反応経路を自動的に探索できる...たった1つの分子をインプットするだけで
すべての反応経路を自動探索
-
- GaussViewhttps://www.hpc.co.jp/chem/software/gaussview/
-
GaussViewは量子化学計算ソフトウェアGaussian専用の可視化ソフトウェアです。分子のモデリング、Gaussianによる量子化学計算の実行、分子軌...Gaussian専用の可視化ソフトウェア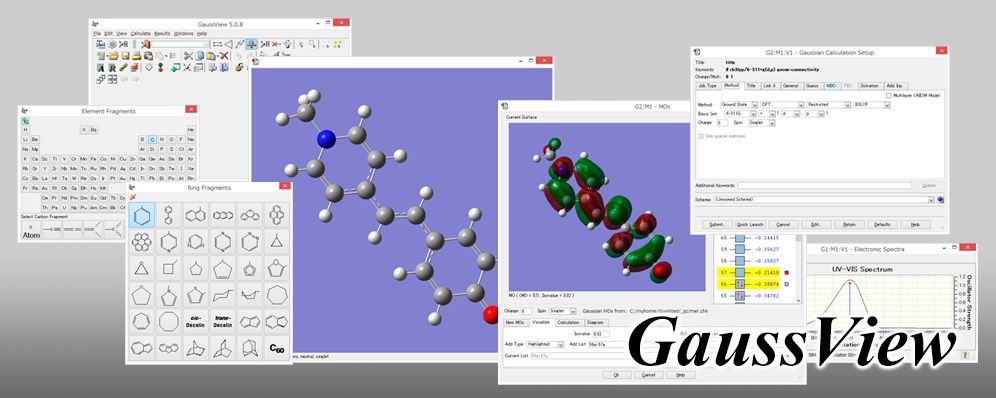
-
- ORCAhttps://www.hpc.co.jp/chem/software/orca/
-
ORCAは、フランク・ネーゼ教授とマックス・プランク研究所の研究グループによって開発された、高性能かつ多機能な量子化学計算プログラムです。分子の電子構造や反...高性能かつ多機能な量子化学計算プログラム
-
- NWChemhttps://www.hpc.co.jp/chem/software/nwchem/
-
NWChemは大規模計算化学の問題を効率的に扱うオープンソース計算化学プログラムです。化学的変換の力学、界面や凝縮相の化学の研究を行うために、米パシフィック...大規模計算化学の問題を効率的に扱う
オープンソース計算化学プログラム
-
- GAMESShttps://www.hpc.co.jp/chem/software/gamess/
-
GAMESSはGaussianと並び広く利用されている、Hartree-Fock(HF)近似を基礎とした原子・分子の非経験的分子軌道法(MO法)・密度汎関数...無償提供の原子、分子の非経験的分子軌道法・
密度汎関数理論計算プログラム
■ 分子動力学
-
- Alphafoldhttps://www.hpc.co.jp/chem/software/alphafold/
-
Alphafoldは、Google DeepMindが開発したタンパク質立体構造予測AIです。アミノ酸配列から三次元構造を高精度に推定できるため、創薬、酵素...Google DeepMindが開発したタンパク質立体構造予測AI
-
- LAMMPShttps://www.hpc.co.jp/chem/software/lammps/
-
LAMMPSは米エネルギー省サンディア国立研究所のS. Plimptonらのグループにより開発された古典分子動力学計算プログラムです。金属や半導体などの固体...古典分子動力学計算の
オープンソース分子動力学計算プログラム
-
- OVITOhttps://www.hpc.co.jp/chem/software/ovito/
-
OVITO(Open Visualization Tool)は、Alexander Stukowski博士によって開発された、主に分子動力学(MD)シミュレ...分子動力学シミュレーション等の
原子・粒子レベルの可視化・解析向け科学ソフトウェア
-
- Gromacshttps://www.hpc.co.jp/chem/software/gromacs/
-
GROMACSはオランダ・フローニンゲン大学で開発された分子動力学(MD)計算プログラムです。地球上で最速のMD計算プログラムを目指しており、MPIあるいは...地球最速のオープンソース
分子動力学計算プログラム
-
- Amberhttps://www.hpc.co.jp/chem/software/amber/
-
Amberは米カリフォルニア大学のP. A. Kollmanらのグループにより開発された、生体分子の分子動力学(MD)計算のための力場群であり、またこれらの...生体分子系のシミュレーションのための
プログラム群
-
- K4https://www.hpc.co.jp/chem/software/k4/
-
K4は神戸医療産業都市推進機構が開発した創薬支援ソフトウェアです。...創薬支援ソフトウェア
■ マテリアルズ・インフォマティクス
-
- M-EVO®https://www.hpc.co.jp/chem/software/m-evo/
-
当社オリジナルのアルゴリズムにより所望の物性を有する多様な分子構造を探索し、提案。複数の目的物性を考慮でき、合成の可能性や溶剤溶解性等の指標も含めたスコアを...所望の物性を有する分子構造の探索を
可能とするクラウドサービス
~データベースレスMIで新たな新素材開発~
-
- AutoMOhttps://www.hpc.co.jp/chem/software/automo/
-
素材開発における分子構造と物性値のデータベースを作成し、csvファイルで保存・M-EVO®の計算データベースとして利用できる計算支援ツールです。...計算化学の課題を解決する
計算支援ツール
■ 固体物理(第一原理計算)
-
- GEAR-Vhttps://www.hpc.co.jp/chem/software/gear-v/
-
化学反応経路の理論的自動探索を世界ではじめて可能にし、触媒設計、材料スクリーニングなどさまざまな分野で活用できるGRRM。結晶や無機固体の非経験的(ab i...GRRMでVASPを使用可能にするインタフェースプログラム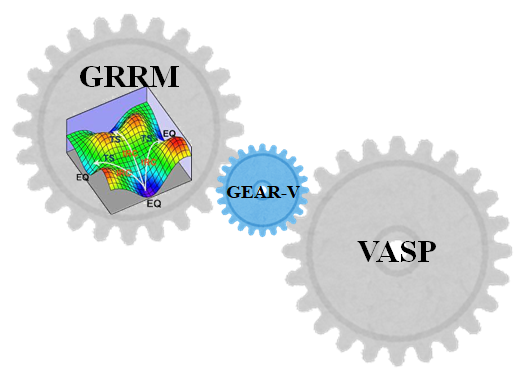
-
- VASPhttps://www.hpc.co.jp/chem/software/vasp/
-
VASPはオーストリア・ウィーン大学で開発された、擬ポテンシャルと平面波基底を用いた非経験的(ab initio)量子分子動力学(MD)計算プログラムです。...ウィーン大学の
固体電子状態計算・バンド計算プログラム
-
- Quantum ESPRESSO (PWscf)https://www.hpc.co.jp/chem/software/quantumespresso/
-
Quantum ESPRESSOは、第一原理の電子構造計算と材料モデリングのためにGNU General Public Licenseとして無料で配布されて...密度汎関数法に基づく
電子状態計算プログラム
■ 旧製品
-
- https://www.hpc.co.jp/chem/software/react1/
-
これまで反応経路を求めるには経験と勘が必要とされ、特に肝となる遷移状態(TS)構造の最適化には大変な苦労を強いられることが常でした。また、せっかくTSが求ま...反応物と生成物自動探索が
Reaction plusに比べ大幅にスピードアップ
-
- Reaction plus Expresshttps://www.hpc.co.jp/chem/software/reactx/
販売終了しています。別途お問い合わせください。 -
Reaction plus Expressは反応経路をたった数分で予測するソフトウェアです。DFTの代わりに半経験的手法を採用し、さらに独自の改良を加えるこ...反応経路をたった数分で予測するソフトウェア
-
- https://www.hpc.co.jp/chem/software/react1n/
-
これまで反応経路を求めるには勘と経験が必要とされ、特に肝となる遷移状態(TS)構造の最適化には大変な苦労を強いられることが常でした。また、せっかくTSが求ま...反応物と生成物を指定するだけで
自動的に反応経路を探索
-
- https://www.hpc.co.jp/chem/software/grrm-basic/
-
たった1つの分子をインプットするだけで、そこから生成されうるすべての生成物やそれを生成しうるすべての反応物、そしてそれらすべての反応経路を自動的に探索できる...たった1つの分子をインプットするだけで
すべての反応経路を自動探索
-
- QM plushttps://www.hpc.co.jp/chem/software/plus_gau2/
販売終了しています。別途お問い合わせください。 -
本ソフトウェアはQMMM plusの機能のうち量子化学計算機能だけに限定した機能限定版となります。特徴についてはQMMM plusを参照ください。...量子化学ソフトウェアで有名なGaussianに
便利な機能をプラス!
-
- GaussRunhttps://www.hpc.co.jp/chem/software/gaussrun/
販売終了しています。別途お問い合わせください。 -
GaussRunはクリックでLinux版のGaussianが操作できます。これが意外と便利! 普段WindowsやMacしか使わないユーザだけでなく、Lin...計算機サーバのGaussianを簡単操作
-
- Solution plus for GROMACShttps://www.hpc.co.jp/chem/software/plus_gmx2/
販売終了しています。別途お問い合わせください。 -
分子動力学ソフトウェアで有名なGROMACS に誰もが簡単に使えるよう機能をプラス!Solution plus for GROMACS
シミュレーションが手軽にできるようになります。...
-
- Solution plus for AMBERhttps://www.hpc.co.jp/chem/software/plus_amb2/
販売終了しています。別途お問い合わせください。 -
分子動力学ソフトウェアで有名なAMBER に誰もが簡単に使えるよう機能をプラス!分子動力学ソフトウェアで有名なAMBERに
誰もが簡単に使えるよう機能をプラス
シミュレーションが手軽にできるようになります。...
-
- QMMM plus 2https://www.hpc.co.jp/chem/software/plus_qmp2/
販売終了しています。別途お問い合わせください。 -
化学計算ソフトウェアで有名なAMBER、GROMACS、Gaussian に誰もが簡単に使えるよう機能をプラス!QM/MM法による溶媒効果を自動計算
シミュレーションが手軽にでき...
Contact
お問い合わせ
お客様に最適な製品をご提案いたします。まずは気軽にお問い合わせ下さい。
075-353-0120
平日9:30~17:30(土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)