
Software
計算化学 ソフトウェア
Reaction plus Pro
販売終了しています。後継のReaction plus Pro2はこちらから
概要
これまで反応経路を求めるには経験と勘が必要とされ、特に肝となる遷移状態(TS)構造の最適化には大変な苦労を強いられることが常でした。また、せっかくTSが求まっても、振動解析してみると反応とは全然関係ないTSだったり、TSからIRC計算が上手く走らず、結局反応経路が求まらないというケースもよくありました。
Reaction plus Proは「研究者のセンス」と「シミュレーション技術」をうまく活用したソフトウェアです。反応物と生成物と指定するだけで自動的に反応経路が求まります。
Proは従来のReaction plusに比べ、大幅にスピードアップ。多くの反応系で4倍以上の速度で反応経路が得られるようになりました。また、量子化学計算エンジンにGaussianを採用したことで、さまざまな量子化学計算条件(DFT汎関数、基底関数、PCM溶媒効果など)に対応。そのほか、路線検索のように経由する中間構造を複数指定することもでき、複雑な反応も手軽に扱えます。
特徴
- 特徴1▸ Reaction plusと比較してProは4倍以上高速になりました。
- 特徴2▸ 遷移状態が手軽に見つかります。
- 特徴3▸ インプットの作成にGaussViewが使えます。
- 特徴4▸ 量子化学計算エンジンにGaussianを採用。さまざまな計算手法が利用できます。
- 特徴5▸ 化学反応の様子がアニメーションで分かります。
- 特徴6▸ よくわかるチュートリアル付き。
- 特徴7▸ サポートオプションもあります。
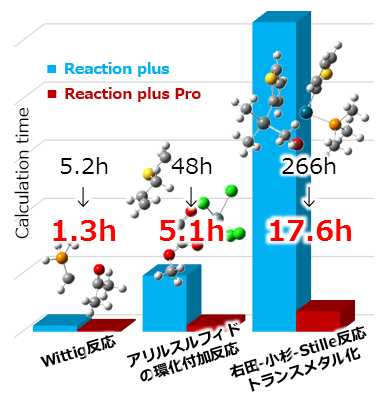
従来比4倍以上高速に!
Reaction plusと比較して、Reaction plus Proは多くの反応系において4倍以上(系によっては10倍以上!)高速に反応経路が計算できるようになりました。8時間かかっていた計算が2時間に、4日かかっていた計算が1日で完了するということ。短期間でさまざまな反応系を調査することができます。
※計算時間は初期構造や計算条件に依存します。
遷移状態が手軽に見つかる!
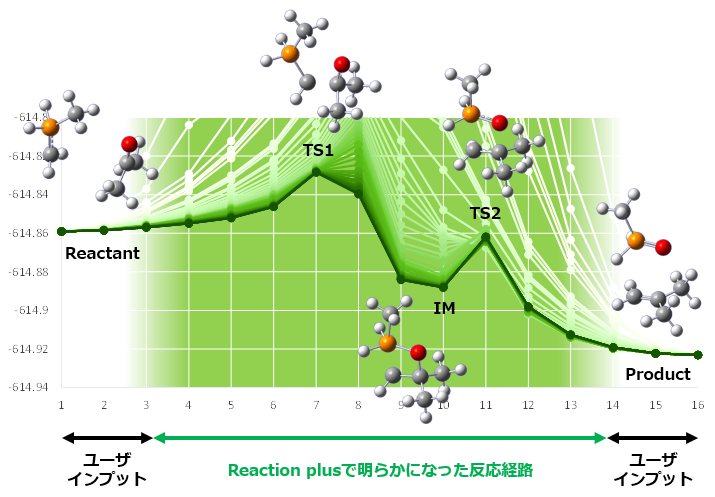
従来の遷移状態最適化法では、これから求めたい遷移状態構造に近い構造を初期構造として指定しなければならず、職人技が要求されました。
Reaction plusではNudged Elastic Band (NEB)法に基づいて反応経路全体を最適化します。指定するのは反応前後の構造だけ。反応経路が自動的に最適化され、経路上の遷移状態構造が苦労せずに求まります。
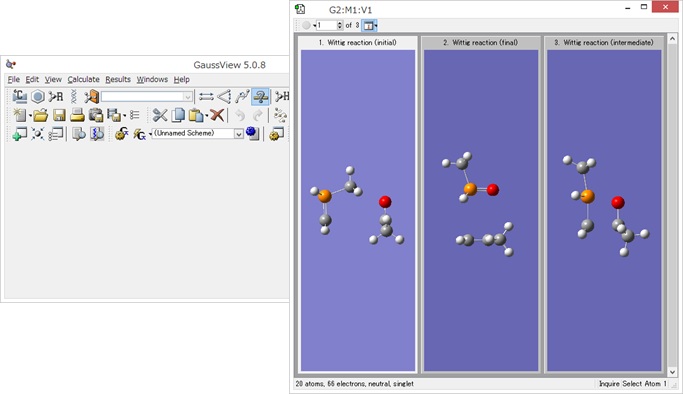
インプットの作成が簡単!
インプットファイルはGaussViewなどの使い慣れた分子エディタで作成することができます。
Reaction plus Proでは経由してほしい途中の構造(中間構造や遷移状態構造の候補)を複数指定することができ、また、分子の向きを自動的に合わせられるなど、Reaction plusにはなかった機能がつきました。
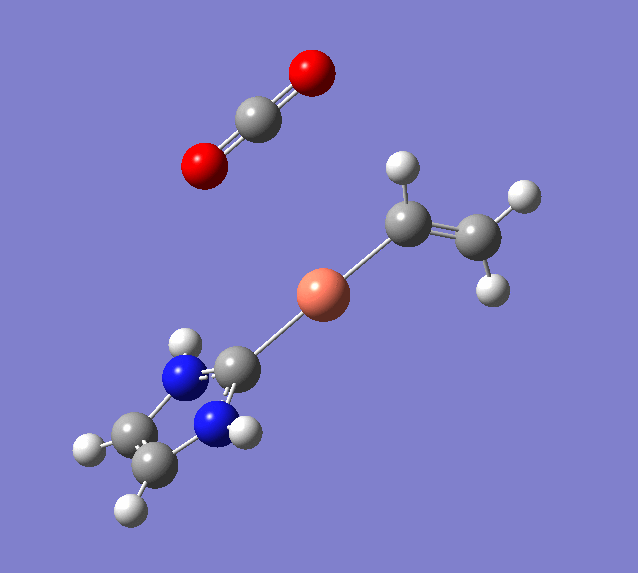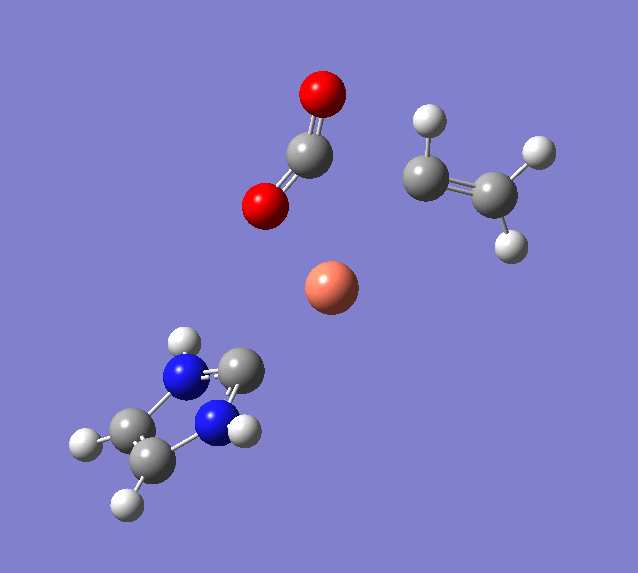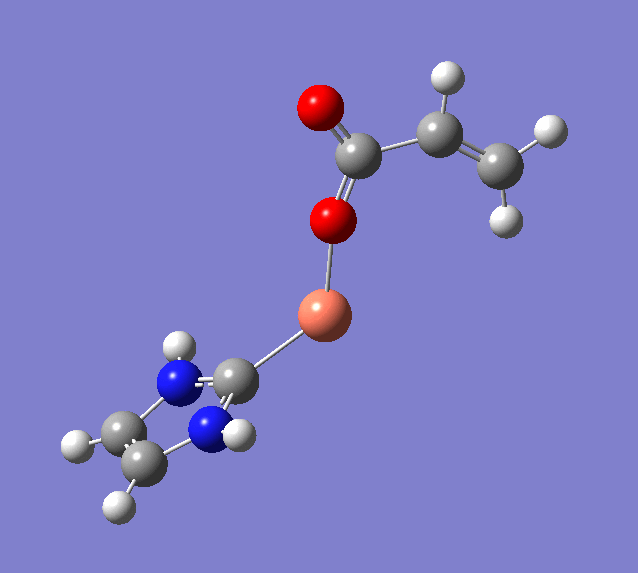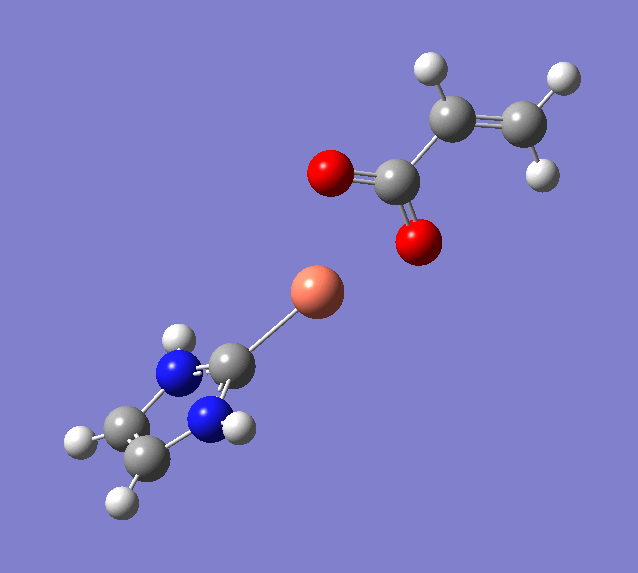
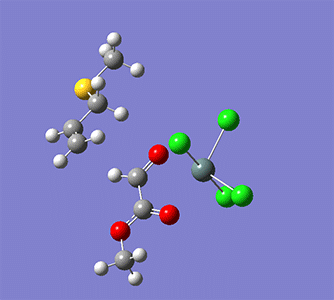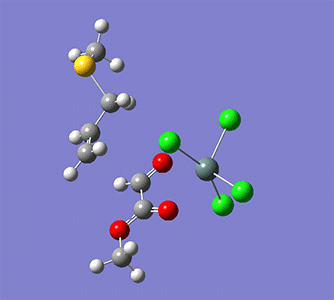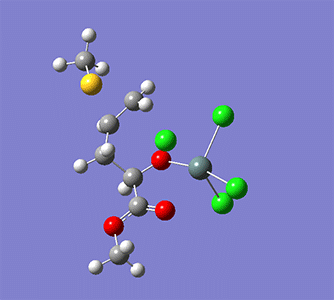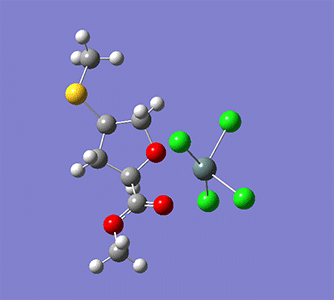
上図では始状態・中間状態・終状態の3構造を指定している。
%nprocshared=16
%mem=16GB
#p B3LYP/6-31G(d) react=(nbeads=16,fit)
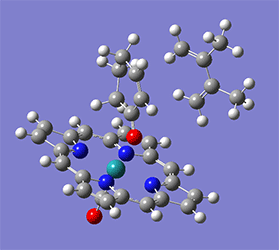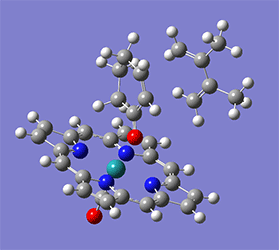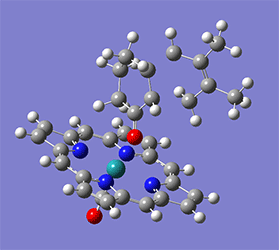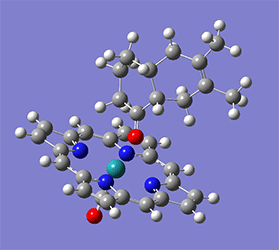
Wittig reaction
0 1
C -2.07681900 -0.18930700 -0.00017300
O -1.94044900 -1.39974600 -0.00140400
C 1.62195100 1.62306900 0.00073100
H 1.25708200 2.03884800 0.93302500
:
作成されるインプットファイルの中身。
赤字部分がReaction plus Proのキーワード。
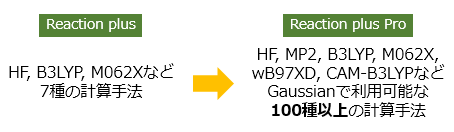
量子化学計算エンジンにGaussianを採用!
量子化学計算エンジンがGaussianになりました。Gaussianに搭載されている多くの計算方法が利用できます。さまざまなDFT汎関数やMP2法が利用できるほか、溶媒効果をPCMで取り入れたり、励起状態での反応経路を調べたりと、反応経路計算の可能性が大きく広がりました。
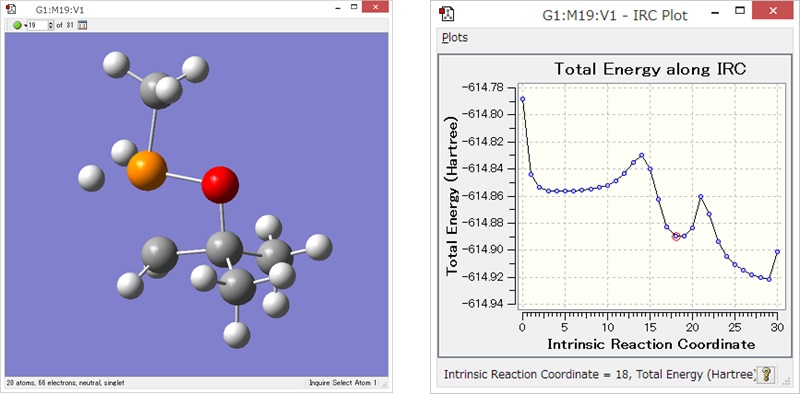
反応の様子がアニメーションでわかる!
Reaction plus ProのアウトプットはGaussian形式とXYZ形式。GaussViewやVMDなどのソフトウェアを使うことで、化学反応の様子をアニメーションで確認することができます。
アニメーションをGIFや動画ファイルとして保存することで、効果的なプレゼンテーションに活用することができます。
よくわかるチュートリアル付き
GaussViewを使ったインプットファイルの作成の仕方や計算の流し方はもちろん、よく使うテクニックなどを解説したチュートリアルが付属します。
サポートオプション(有償)もあります
「テクニックが知りたい」「エラーを解決したい」など、実際に使っていて出会う様々な問題に弊社スタッフが対応いたします。メールでの質問対応のほか、出張による対面での対応が可能です。
Reaction plusの仕組み
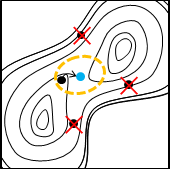
従来の遷移状態(TS)最適化では、TS構造が分からないにもかかわらず、TS構造に近い構造を初期値に指定する必要がありました。
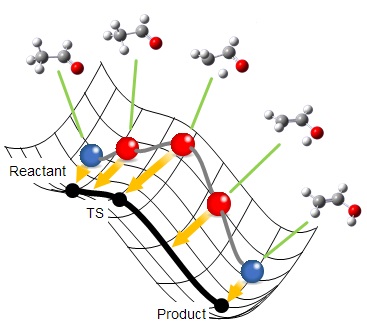
Reaction plusでは反応経路全体を最適化するNEB法を採用しています。
分子構造を1つのビーズに例えると、反応経路はビーズがひもで連なったものとして表すことができます。このひもを反応ポテンシャル曲面上でコロコロと転がしていくと、各ビーズがエネルギーが低くなる方向へ転がろうとする力とひもの束縛力のバランスにより、最終的にひもの形は解となる反応経路に収束します。これが反応経路最適化の基本的な仕組みです。
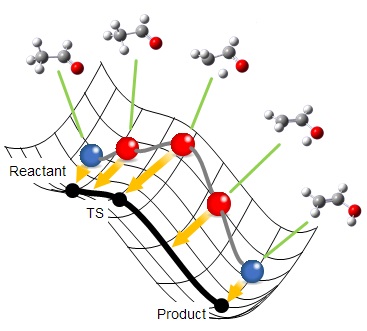
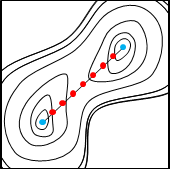
まず、始状態・終状態の構造をユーザが与えると、その間に等間隔にビーズ(分子構造)が配置されます。
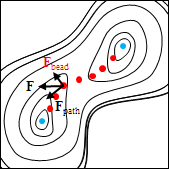
各ビーズに働く力に従ってビーズを最適化していくと、ビーズは始状態ないし終状態に向かって移動しようとしますが、隣り合うビーズが近づかないよう、ひもと直交する方向成分に動かすことによって、目的の正しい反応経路に近づいていきます。
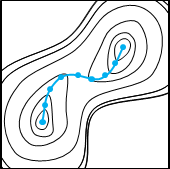
ビーズの動きがなくなると、最適化は完了です。各ビーズに働く力はひもの方向、すなわち、反応経路に沿った方向のみであることから、正しく経路が求まっていることがわかります。
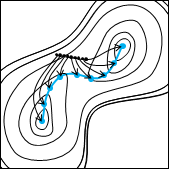
初期値として与える両端の分子構造は、安定構造である必要はありません。適当に初期値を与えても、反応経路が求まります。
「こんな反応機構はどうだろうか」とユーザが感覚的に与えた構造からも反応経路を求めることができ、これまでの研究の経験とセンスが活かせます。
計算例
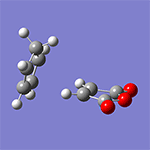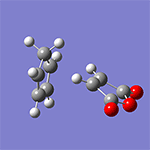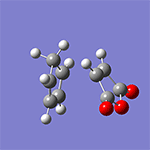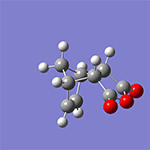
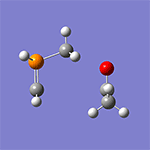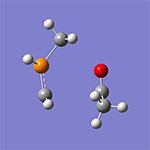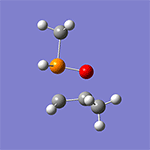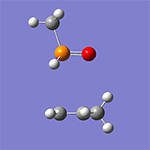
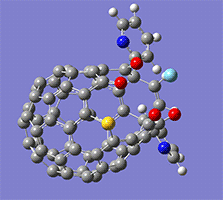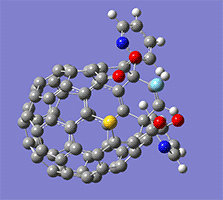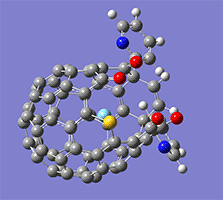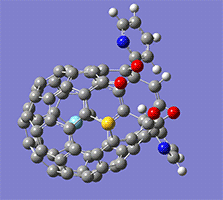
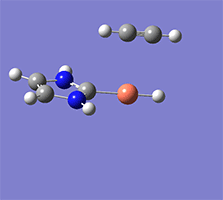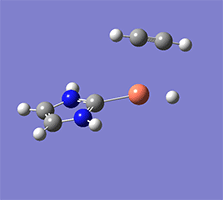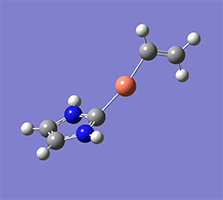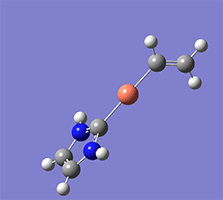
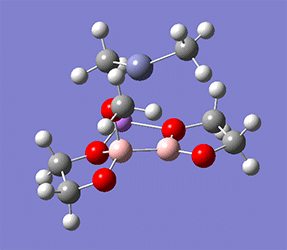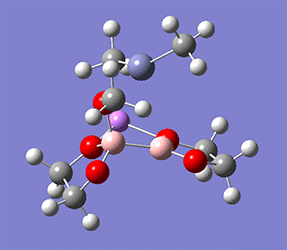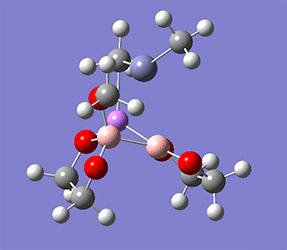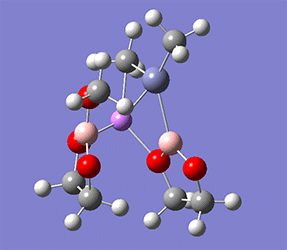
仕様・価格
仕様
| 製品名 | Reaction plus Pro ver. 1.0 for Linux | Reaction plus Pro ver. 1.0 for Windows |
|---|---|---|
| 反応経路最適化 | ○ | |
| 指定可能な中間構造の数 | 任意の数 | |
| 初期構造の座標軸の自動修正 | ○ | |
| 計算結果を再利用した 最適化計算 | ○ | |
| 量子化学計算手法 | GaussianのOpt機能に対応するすべての計算手法 | |
| 基底関数 | Gaussianで利用可能なすべての基底関数 | |
| GUIによるインプットの作成 | GaussViewにて可能 ※1 | |
| アニメーション表示 | GaussView、VMDにて可能 ※1 | |
| 最大並列数 | 1ノード搭載コア数 ※2 | |
| 同梱物 | ソフトウェア本体、マニュアル、チュートリアル | |
| 動作環境 ※3,4 | OS: Red Hat Enterprise Linux 6 以降 または CentOS 6 以降 ソフトウェア: Gaussian 09 Rev. C01以降 | OS: Windows 10, Windows 8.1, Windows 7, Windows Server 2012 R2, Windows Server 2012, Windows Server 2008 R2 (いずれも64bit) ソフトウェア: Gaussian 09 Rev. C01以降 |
| 発売日 | 2016年1月1日 | 2016年4月18日 |
※ 表示価格は税抜価格です。
※ 保証期間は購入後1年間となります。ソフトウェアの活用方法に関するサポートは別途「Reaction plus 使い方サポート」をご利用ください。
※1 GaussView 5.0.8 for Windows, VMD 1.9.1にて動作確認を行っております。
※2 ノード間並列には対応していません。
※3 本ソフトウェアの動作には、別途Gaussian09が必要です。
※4 すべての環境についての動作を保証するものではありません。特に、セキュリティソフトが常駐しているWindows環境で計算を行う場合、セキュリティソフトの動作により計算が停止することがあります。
価格
| Reaction plus Pro ver. 1.0 ( for Linux / for Windows ) |
|---|
| 200万円 / 1ノード (アカデミック価格:60万円) |
※ アカデミックパッケージに機能制限はありません。ただし、研究成果を論文等で公開の際には本ソフトウェアを使用した旨を記載してください(詳細はこちら)。
サポートオプション
| Reaction plus 使い方サポート | ||
|---|---|---|
| メールサポート | 出張サポート | |
| 内容 | 質問メール対応(ひと月あたり4回) + 使い方セミナー受講(初回のみ) ※1 | 対面での質問対応 ※1 |
| 価格 | 80万円/年 | 25万円/日 (10時~18時) |
※1 遠隔地での実施の場合は別途出張費が発生することがあります。
旧製品との機能比較
| 製品名 | Reaction plus Pro ver. 1.0 (2016年1月1日 発売) | Reaction plus ver. 1.0 (2015年5月1日 発売) |
|---|---|---|
| 指定可能な中間構造の数 | 任意の数 | 1構造まで |
| 初期構造の座標軸の自動修正 | ○ | × |
| 計算結果を再利用した 最適化計算 | ○ | × |
| 量子化学計算エンジン | Gaussian09 | NWChem 6.5 |
| 量子化学計算手法 | GaussianのOpt機能に対応するすべての計算手法 | HF, B3LYP, M06-2Xなど7種類 |
| 基底関数 | Gaussianで利用可能なすべての基底関数 | 6-31G、cc-pVDZなど24種 |
| 最大並列数 | 1ノード搭載コア数 | |
※ 旧製品 Reaction plus についてはこちらをご覧ください。
動作報告
for Linux
| 計算機サーバ OS | Gaussian | |
|---|---|---|
| ○ | CentOS 6.7 | Gaussian09 Rev.E01 |
| ○ | CentOS 6.5 | Gaussian09 Rev.D01 |
| ○ | CentOS 6.4 | Gaussian09 Rev.D01 |
| ○ | CentOS 6.4 | Gaussian09 Rev.C01 |
| クライアント OS | アプリケーション (アニメーション表示) | |
|---|---|---|
| ○ | Windows 8.1 | GaussView 5.0.8 (logファイル) |
| ○ | Windows 8.1 | VMD 1.9.1 (xyzファイル) |
| △ | Windows 8.1 | Avogadro 1.1.1 (xyzファイル・logファイル) ※ただし、logファイルについては動作しないことがある |
| ○ | Windows 7 | GaussView 5.0.8 (logファイル) |
for Windows
| OS | Gaussian | |
|---|---|---|
| ○ | Windows 10 | Gaussian09 Rev.E01 |
| ○ | Windows 8.1 | Gaussian09 Rev.E01 |
| ○ | Windows 8.1 | Gaussian09 Rev.D01 |
| ○ | Windows 7 | Gaussian09 Rev.E01 |
| ○ | Windows 7 | Gaussian09 Rev.D01 |
| ○ | Windows 7 | Gaussian09 Rev.C01 |
リリースノート
- version 1.0
- version 1.0.0
- 初版。
- version 1.0.1
- Windows環境への対応。
- スクラッチファイル名の衝突を避けるため、スクラッチファイルをworkディレクトリ内で作成するように変更。
- version 1.0.2
- GaussView 6で閲覧時の不具合を修正。
- ライセンスエラー時の挙動を修正(Windows版)。
※ 本ソフトウェアの内容は予告なく変更することがあります。あらかじめご了承ください。
※ 記載された各社名・各製品名・各ロゴ等は、各社の登録商標または商標です。
サポート情報
よくある質問
ご購入前の質問
- Reaction plusとReaction plus Proの違いを教えてください。
- Proは従来のReaction plusに比べ、非常に高速であることが大きなメリットです。その他、Gaussianに搭載されているほとんどの計算方法が使えることや、インプットに複数の中間構造を指定することで複雑な反応に対応できる点も有用です。
一方、従来のReaction plusの優位点は費用が安く抑えられることにあります。また、動作要件としてGaussianは不要のため、Reaction plus単体の費用で計算を始めることができます。
- アカデミックパッケージに機能制限はありますか?
- ソフトウェアの機能において制限はありません。ただし、研究成果を論文等で公開の際には本ソフトウェアを使用した旨を記載してください。
(Reaction plusを使ってTS候補構造を求め、それを初期値にしてソフトウェアAAAによるTS計算を行った場合)
[2] H. Jónsson, G. Mills, K. W. Jacobsen, Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions, in Classical and Quantum Dynamics in Condensed Phase Simulations, Ed. B. J. Berne, G. Ciccotti and D. F. Coker, 385 (World Scientific, 1998); G. Henkelman and H. Jónsson, Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points, J. Chem. Phys. 113, 9978 (2000)
- Gaussian 16には対応していますか?
- 対応しています。動作検証の結果、正常に動作することが確認されました。
- カタログはありますか?
- カタログや計算事例集は、こちらのページからダウンロードできます。
- 体験版はありますか?
- はい。期限付のライセンスを発行させていただいております。詳細につきましては、お問い合わせ下さい。
- リース契約は可能ですか?
- 通常は買い取りにてのご購入となっておりますが、お客様のご都合に合わせて提案することは可能です。お問い合わせください。
- サポートはありますか?
- 有償サポートをご用意しております。詳細はこちらをご覧ください。
- どのくらいのスペックの計算機が必要ですか?
- CPUコア数を重視した計算機構成をおすすめします。参考に、計算例のWittig反応(16ビーズ)では、Intel Xeon E5-2690 2.90GHz×2(16コア)搭載の計算機で計算時間は1時間19分(最適化サイクル=62回)でした。
- GUI(グラフィカルユーザインターフェース)は付属しますか?
- 付属しません。本ソフトウェアはコンソールアプリケーションです。インプットファイルの作成や化学反応の可視化については、GaussViewなどのソフトウェアをご利用ください。
- バージョンアップは有償ですか?
- マイナーバージョンアップに関しましては、保証期間内に限り無償でアップデート可能です。アップデート方法につきましては、リリース時にメールにてご案内いたします。
Reaction plusの機能に関する質問
- ONIOMは使えますか?
- 今のところ未対応です。
- 溶媒効果を入れた計算はできますか?
- PCMなど、Gaussianが対応している溶媒効果モデルで計算ができます。
- 原子電荷を見ることはできますか?
- GaussViewにて Results → Charge Distribution… より表示することができます。(本機能は実験的機能のため、予告なく変更されることがあります。現バージョンではNPA電荷を計算した場合であっても、その値をMulliken電荷として表示します。)
- GRRMとの違いについて教えてください。
- 本ソフトウェアでは狙った1つの反応経路を最適化するため、計算速度は非常に速く、比較的大きな分子の計算も可能です。一方、GRRMではすべての反応経路を全探索するため、人間業では対応の難しいきわめて複雑な反応経路の探索に向いています。その反面、計算コストが莫大となるため、大きな分子の経路探索には大型計算機が必要となります。
トラブルシューティングに関する質問
- 計算が走らないのですが?
- a) Reaction plusおよびGaussianが正しくインストールされていることを確認してください。
b) プログラムのインストール先やインプットファイルのパスに、全角文字や半角カナ等が含まれないようにしてください。
c) ライセンスファイルが正しいディレクトリに置かれていることを確認して下さい。
- 実行直後に終了してしまうのですが?
- a) ルートセクションのキーワードが正しく与えられているか、再度ご確認ください。
b) フォーマットを確認してください。タイトル→座標→タイトル→座標→…のようにタイトルと座標を交互に入力する必要があります。
c) インプットとして3構造以上を指定する場合は、NGeomキーワードを指定してください。
- 計算中にエラー終了してしまうのですが?
- a) 計算手法や分子構造が適切ではないと、エラーとなることがあります。
b) Gaussianのスクラッチディレクトリにファイルがたまっている状態では、ファイル名が被り、エラーとなることがあります。スクラッチディレクトリを空にしてください。
c) 本プログラムは、始状態・終状態の間に遷移状態があることを前提としています。エネルギーポテンシャルを単調に登るだけ、下るだけの反応の場合、正しく動作しないことがあります。
d) 入力した始状態・中間状態・終状態の間を線形に補間してビーズを生成させています。補間の際に原子間距離が近づきすぎるなど構造がおかしいとき、正しく動作しないことがあります。
e) セキュリティソフトの影響で計算が停止することがあります。
特に、Windows版をGaussian 09 Revision C.01のソフトウェア環境で利用した際に動作不良となるケースが報告されています。Gaussianスクラッチファイルの拡張子scrがスクリーンセーバーに関連付けられている場合、この関連付けを解除することで不具合を回避できる可能性があります。(なお、Revesion D.01、 E.01ではスクラッチファイルの拡張子はskrに変更されています。)
- 途中で計算を停止させたいのですが?
-
for Linux
a) コマンドラインにて実行させた場合は、計算停止プログラムkilltreeを使用して停止させてください。
b) ジョブスケジューラを使用して実行させた場合は、ジョブ停止コマンド(bkillやqdel)を使用して停止させます。
c) 手動で停止させる場合は、reactg → g09 → l???.exeの順に停止させてください。 - for Windows
a) kill-reactg-g09.batをダブルクリックしてください。停止処理が完了すると、自動的にコマンドプロンプトウィンドウが閉じます。(※ kill-reactg-g09.batはWindowsバッチファイルです。reactg.exe,g09.exe,l502.exeなどのプログラムを強制停止します。環境に合わせて編集して使用してください。)
b) 手動で停止させる場合は、タスクマネージャーより、reactg → g09 → l???.exeの順に停止させてください。
- 計算結果が思わしくありません。
- a) 始状態・終状態の構造が最適化構造(または、反応経路上の構造)ではないとき、正しく求まらないことがあります。
b) 始状態・終状態の構造だけを与えると、反応経路初期値として始状態と終状態を直線的に結ぶ経路が選ばれますが、そのような経路が初期値として不適切である可能性があります(例えば、cis-スチルベンからtrans-スチルベンへの異性化反応など)。その場合は、3点目として中間状態を適切に指定してやる必要があります。
c) 始状態・終状態・中間状態の分子構造において原子のラベル番号の対応が適切でないと、不適当な反応経路や遷移状態構造を与えることがあります。
d) 始状態・終状態・中間状態の座標軸が適切でないと、不適当な反応経路や遷移状態構造を与えることがあります。FitAxisオプションを利用するか、手動で座標軸を合わせてください。
e) インプットの分子構造を始状態→中間状態→終状態の順に指定していませんか? 正しくは、始状態→終状態→中間状態の順です。
f) 本プログラムではビーズの動きが十分小さくなったと判断した時に収束したとみなし最適化が終了しますが、化学的な観点からは収束が十分でないことがあります。その場合は、調べたい構造(例えば遷移状態構造)前後の構造を両端構造として再計算するなど、局所的な反応経路の精査を行ってみてください。
g) 本ソフトウェアは短時間で反応経路を求めることを目的としているため、厳密な安定性解析は行っておりません。精確な遷移状態を得たい場合はReaction plusで求まった構造をもとに、Gaussianなどの量子化学計算ソフトウェアにて遷移状態最適化計算やIRC計算を行ってください。
その他の質問
- Reaction plus Proを分子研のスパコンで使うことはできますか?
- 従来版のReaction plusは分子研で無償利用できますが、Proは利用できません。



お問い合わせ
平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)