
Software
計算化学 ソフトウェア
販売終了しています。別途お問い合わせください。
Solution plus for AMBER
販売終了しています。別途お問い合わせください。
分子動力学ソフトウェアで有名なAMBER に誰もが簡単に使えるよう機能をプラス!
シミュレーションが手軽にできるようになります。
特徴
1. MDシミュレーションがとにかく簡単!
MDシミュレーションは、分子を作成し、分子を配置して系を作成し、平衡化して、トラジェクトリを解析し…と多くの手続きからなっています。この複雑さゆえ、専門の研究者ですら1日がかりの作業になることが多々ありました。
本ソフトウェアでは溶液中の各分子構造ファイルとちょっとした設定だけで計算を始めることができます。分子の配置からMD計算、そして解析計算まですべて自動化! 待っているだけで答えが得られます。
2. 計算結果が早くわかる!
計算自動化の恩恵は時間に現れます。手作業で行う場合、あれこれと試行錯誤することが多く、結果的に1日に評価できる材料・解析項目は限られてしまいます。
本ソフトウェアを使用すれば、材料の種類や濃度を変えた計算を次々と実行することができます。例えば、1日で480の電解質材料の評価※が可能です。
※ Intel Xeon E5-2690 2.90GHz x2(16コア)計算機10台の構成で実行時。
3. 混合溶媒中の物性が手軽に評価できる!
計算の設定は分子構造ファイル(PDB/Mol2/Gaussian形式)を用意して、分子数を指定するだけ。これだけで分子を適度に混ぜ合わせた系のシミュレーションが実行できます。
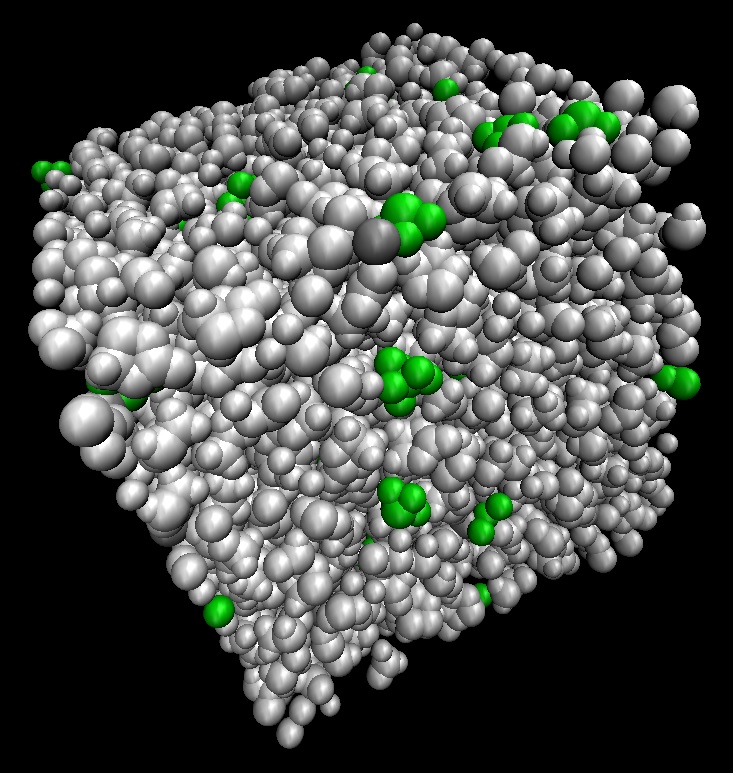
4. 溶液温度による違いがわかる!
本ソフトウェアでは、実際の温度・圧力環境でシミュレーションができるため、温度を変えて比較計算が可能です。
また絶対零度・静止状態の量子化学計算を超えて運動状態が扱えるため、エネルギーなどの物性情報を平均値と標準偏差といった統計量として得ることができ、現象理解の可能性を広げました。
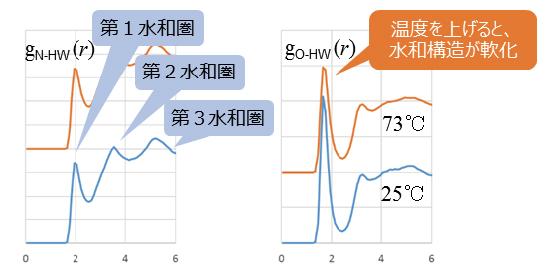
5. AMBERがもっと便利に!
分子動力学シミュレーションの自動化を実現したプログラムのほか、AMBERトラジェクトリの計算解析ツールが用意されています。
機能紹介
① MD自動計算
自動計算
溶液系の計算を始めるには、分子の作成、力場の設定、溶媒分子の配置など多くの手続きを、さまざまなプログラムを駆使して準備する必要がありました。
Solution plus for AMBERではこれらの手続きをすべて自動化。分子構造ファイルと温度や圧力といった溶媒環境情報を準備するだけで計算を始めることができます。ユーザは計算が終わるまで待っているだけでよく、溶液中のエネルギーやスペクトルなどの物性が自動的に計算されます。

リスタート機能
計算手続きの途中から実行することが可能です。
こんな時に便利です
- ①中断した計算を再開したいとき
- 他に優先させたい計算ができたときや、計算機のトラブルなどで、計算を中断させてしまった場合でも、正しく計算された部分の続きから計算を再開させることができます。
- ②同じような分子系を評価するとき
- 分子数が異なるだけの計算の場合、分子の力場設定ディレクトリを残した状態で計算を始めれば、力場設定処理が省略され、指定分子数で溶媒分子配置以降の処理が自動実行されます。
② MD解析
エネルギーや温度などの時間変化や統計値
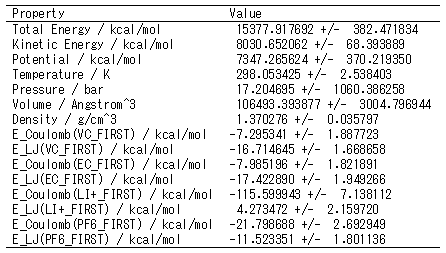
系全体のエネルギーや温度、密度などの時間変化を見ることができます。溶質分子と溶媒分子との間の相互作用エネルギーの時間変化もわかります。
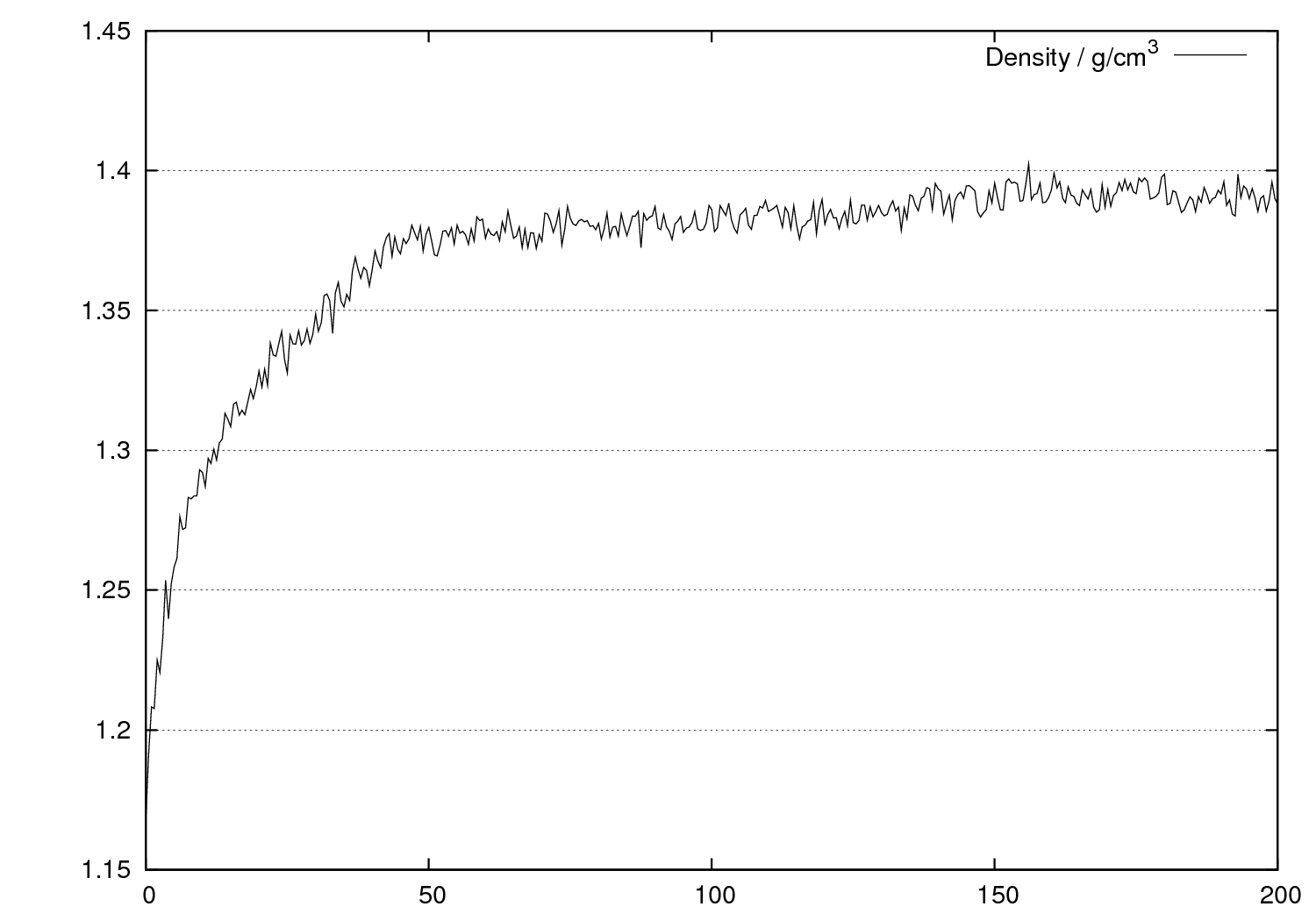
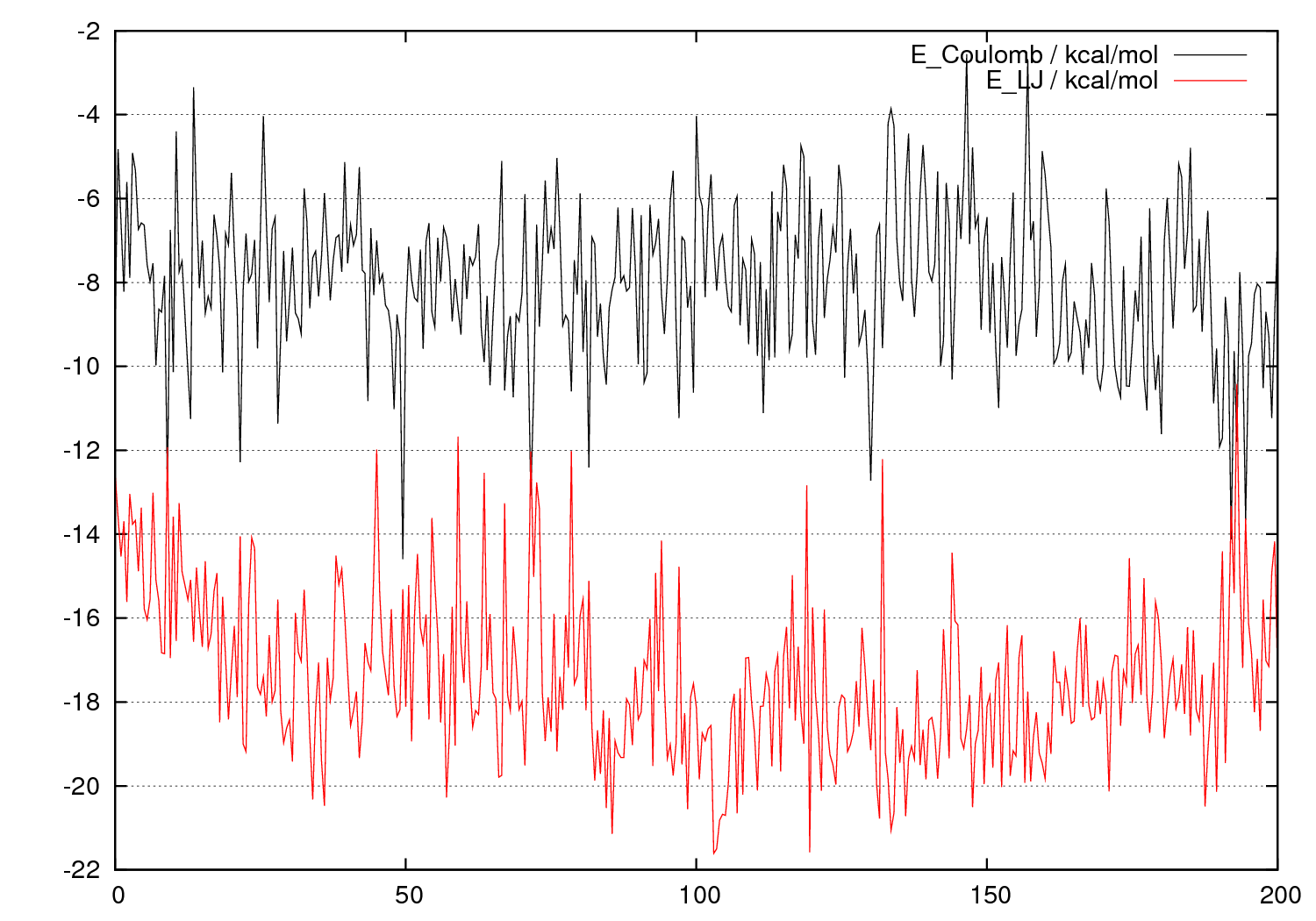
そのほか、統計量(平均値と標準偏差)も出力されます。
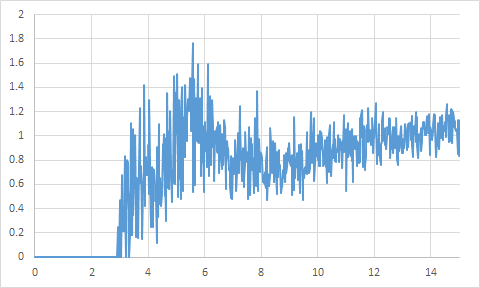
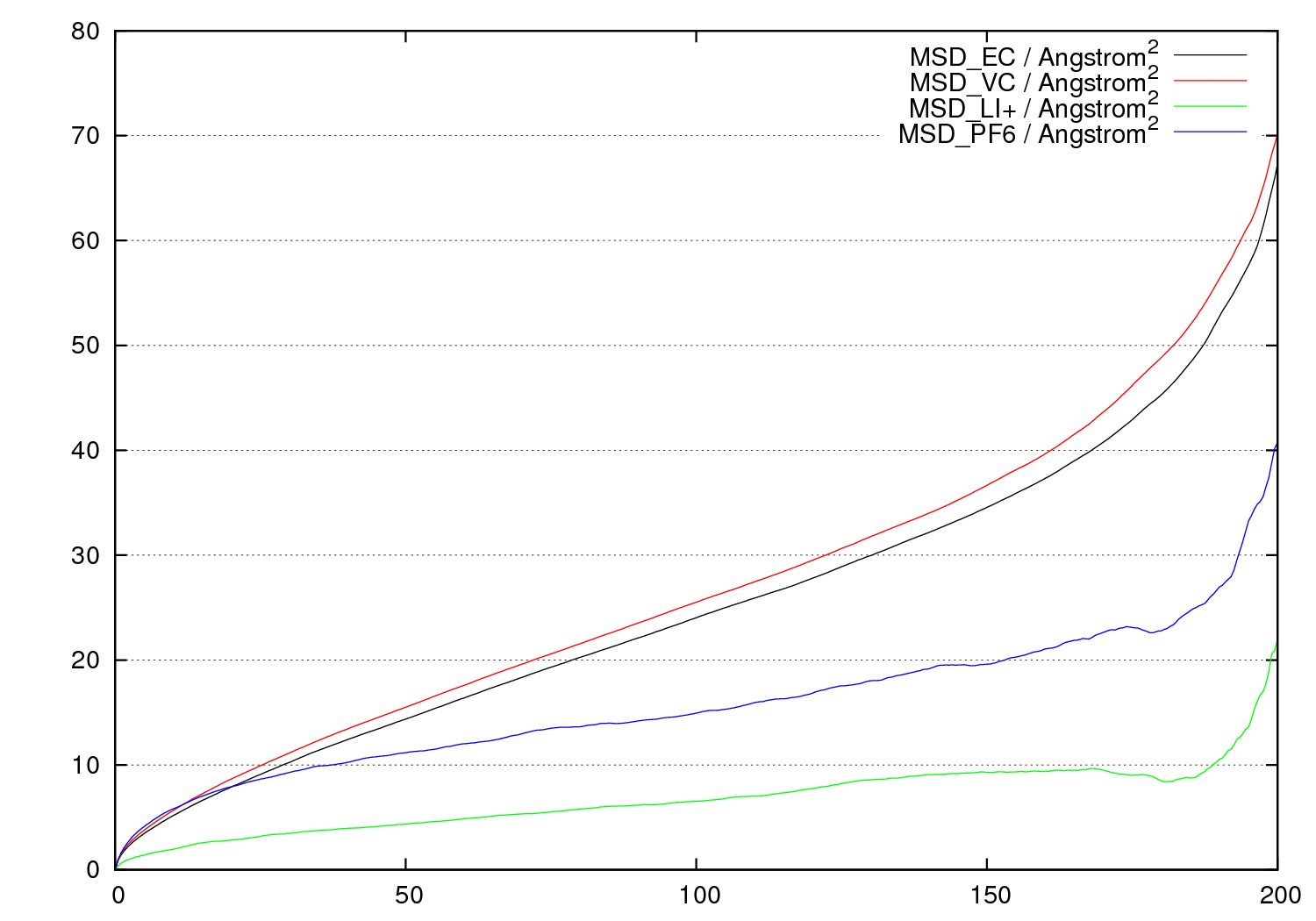
溶媒の分布や拡散の評価
MDのトラジェクトリファイルから溶媒の分布や拡散挙動が解析できます。
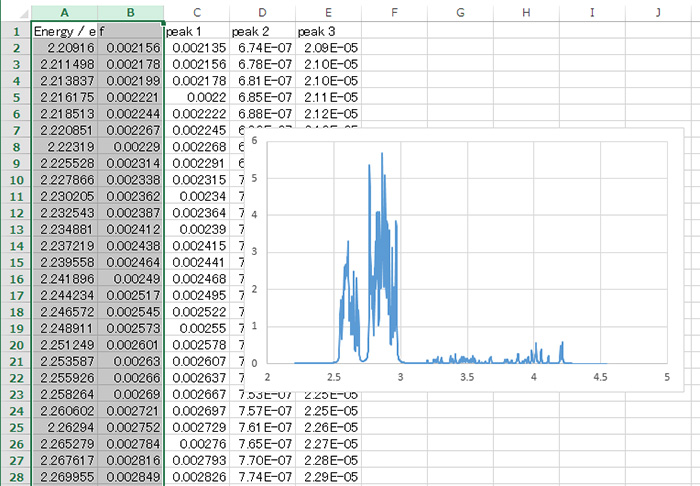
③ レポート出力
HTMLレポートとテキストレポート
計算結果はHTMLファイルにまとめられるので、ブラウザで結果を一覧することができます。タブ区切りの数値データファイルは同じ場所のdataディレクトリにまとめて保存されます。
HTMLファイルのほかテキストファイルにも同様の内容が出力されますので、GUIの利用できない環境でも計算結果の確認が可能です。
使い方
MD自動計算プログラム SOLUTION の使い方を説明します。
1. 分子を作成します
溶質分子や溶媒分子はGaussViewなどで使い慣れたツールで作成することができます。
2. 計算条件を設定します
分子構造ファイルと分子数を指定し、温度などの計算条件を指定するだけで、設定完了です。
MD計算に詳しい方であれば、MD計算過程の細かい指定も可能です。

3. 計算を実行します
コマンドはたった1行。あとは計算が終わるまで待つだけです。
![]()
計算結果はHTML形式で出力されますので、ブラウザで一覧することができます。
仕様・価格
![]()

構成内容
| 機能 | ツール名 | 説明 |
|---|---|---|
| MD自動計算 | SOLUTION | MD計算や量子化学計算を活用した溶液シミュレーションの実行 |
| MD解析 | AMBINFO | AMBERのアウトプットファイルから情報を抽出 |
| レポート | - | 解析結果を成形して出力 (SOLUTION, AMBINFOに装備) |
動作環境
Red Hat Enterprise Linux 6.x または CentOS 6.x、および、下記ソフトウェア
| ソフトウェア | |
|---|---|
| Python 2.6 | ◎ |
| LSF, LAVA, Grid Engine のいずれか※1 | ○ |
| Gaussian09※2 | ○ |
| AMBER14※3 | ○ |
| AmberTools14 | ◎ |
| GROMACS 4.6または5.0 | ◎ |
| acpype.py | ◎ |
| Open Babel 2.x※4 | ◎ |
| Gnuplot※5 | ○ |
| Image Magick※5 | ○ |
◎=必須 ○=推奨
※1 複数ノードで計算する際に必要になります。
※2 力場処理(原子電荷)の精度が向上します。
※3 古典MD計算が高速化されます。
※4 Open Babel 2.2.3にて動作確認しています。
※5 グラフ画像の自動生成をする際に必要になります。gnuplot 4.2 patchlevel 6, ImageMagick 6.5.4-7 にて動作確認しています。他のバージョンでは正常動作しないことがあります。
価格
| Solution plus for AMBER |
| 80万円 |
※ 表示価格は税抜価格です。



お問い合わせ
平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)