
Software
計算化学 ソフトウェア
販売終了しています。別途お問い合わせください。
QMMM plus 2
販売終了しています。別途お問い合わせください。
化学計算ソフトウェアで有名なAMBER、GROMACS、Gaussian に
誰もが簡単に使えるよう機能をプラス!
シミュレーションが手軽にできるようになります。
特徴
1. MDシミュレーションがとにかく簡単!
MDシミュレーションは、分子を作成し、分子を配置して系を作成し、平衡化して、トラジェクトリを解析し…と多くの手続きからなっています。この複雑さゆえ、専門の研究者ですら1日がかりの作業になることが多々ありました。
本ソフトウェアでは溶液中の各分子構造ファイルとちょっとした設定だけで計算を始めることができます。分子の配置からMD計算、そして解析計算まですべて自動化! 待っているだけで答えが得られます。
2. 計算結果が早くわかる!
計算自動化の恩恵は時間に現れます。手作業で行う場合、あれこれと試行錯誤することが多く、結果的に1日に評価できる材料・解析項目は限られてしまいます。
本ソフトウェアを使用すれば、材料の種類や濃度を変えた計算を次々と実行することができます。例えば、1日で480の電解質材料の評価※が可能です。
※ Intel Xeon E5-2690 2.90GHz x2(16コア)計算機10台の構成で実行時。
3. 混合溶媒中の物性が手軽に評価できる!
計算の設定は分子構造ファイル(PDB/Mol2/Gaussian形式)を用意して、分子数を指定するだけ。これだけで分子を適度に混ぜ合わせた系のシミュレーションが実行できます。
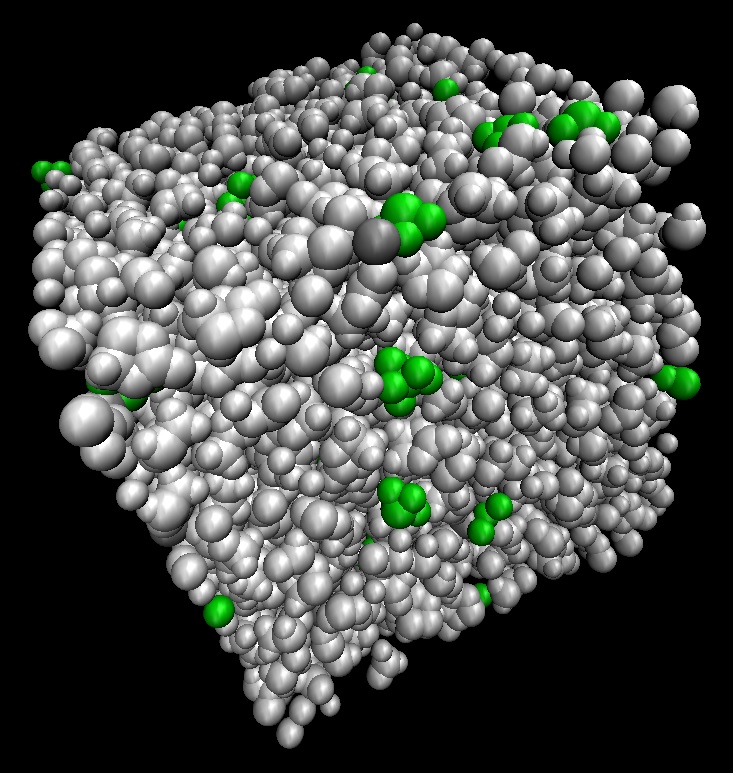
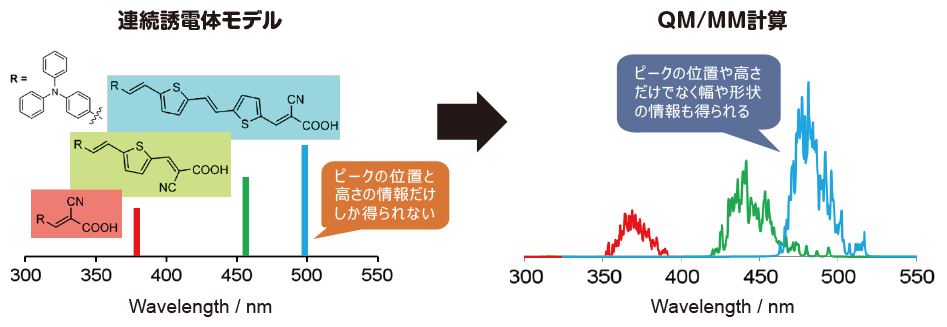
4. 誘電体モデルでは説明がつかなかった溶媒効果がわかる!
量子化学計算では通常、計算したい対象系を絶対零度・誘電体中の問題にモデル化して計算を行います。しかしながらこのモデル化が原因で期待した結果が得られないことが多々ありました。
本ソフトウェアでは着目している分子の周囲に溶媒を分子として配置した計算を行います。水素結合など局所的な相互作用が取り込まれ、誘電体モデルを超えた精度で溶液中の構造安定性、溶媒の配向状態、IR・Raman・UV-Vis・CD・NMRスペクトルがわかります。
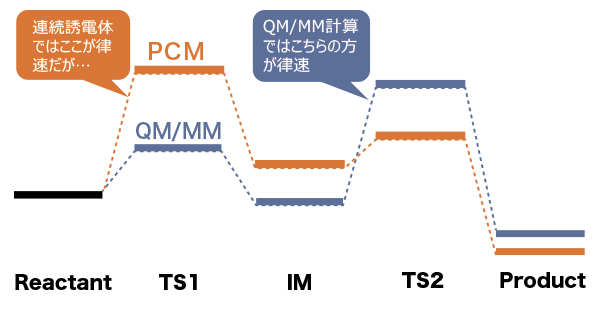
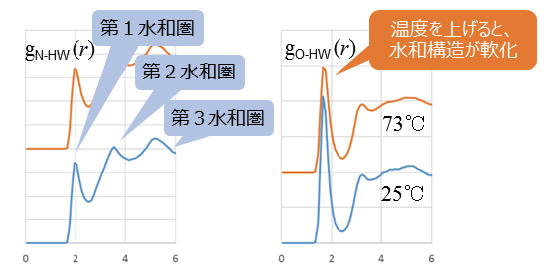
5. 溶液温度による違いがわかる!
本ソフトウェアでは、実際の温度・圧力環境でシミュレーションができるため、温度を変えて比較計算が可能です。
また絶対零度・静止状態の量子化学計算を超えて運動状態が扱えるため、エネルギーなどの物性情報を平均値と標準偏差といった統計量として得ることができ、現象理解の可能性を広げました。
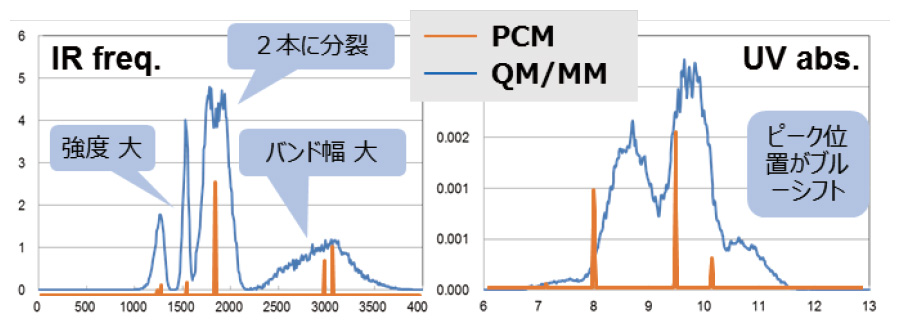
6. 溶液中のスペクトルがわかる!
孤立系の量子化学計算では、振動スペクトルや吸収スペクトルは線スペクトルでしか得られませんでした。
溶液中の分子ダイナミクスを評価することで、実験値に直接対応する幅を持ったスペクトルが得られます。
7. AMBER, GROMACS, Gaussianがもっと便利に!
本ソフトウェアでは分子動力学ソフトウェアで有名なAMBER, GROMACS、量子化学ソフトウェアで有名なGaussianを採用しています。
化学シミュレーションの自動化を実現したプログラムのほか、計算解析ツール、一括編集ツールなど、AMBER, GROMACS, Gaussianを単体のプログラムとして使用した際にも利用できるツールが用意されています。
Version 2.0の新機能
- 複数種類の分子の混合溶媒が扱えるようになりました。
- Gaussianユーザ向けの機能が充実しました(一括編集機能や統計解析機能など)。
- レポート機能が追加されました。
- 使用するコア数に上限を設定できるようになりました。
- ジョブ管理ソフトを使用しない並列・並行計算が可能になりました。
- 計算のリスタート機能が追加されました。
- AMBER14がインストールされていない環境(AmberTools14のみがインストールされている環境)に対応しました。
- sanderを機能拡張し、任意の量子化学計算ソフトウェアに対応しました。
機能
① MD自動計算
自動計算
溶液系の計算を始めるには、分子の作成、力場の設定、溶媒分子の配置など多くの手続きを、さまざまなプログラムを駆使して準備する必要がありました。
QMMM plusではこれらの手続きをすべて自動化。分子構造ファイルと温度や圧力といった溶媒環境情報を準備するだけで計算を始めることができます。ユーザは計算が終わるまで待っているだけでよく、溶液中のエネルギーやスペクトルなどの物性が自動的に計算されます。

計算スタイルを選択するだけでシミュレーションの設定が自動的に行われます。もちろん、平衡化MDを高速なGROMACSで行い、ab initio QM/MMによるサンプリングMDをAMBER+Gaussianで実行するなど、計算手続きを細かく指定することもできます。
リスタート機能
計算手続きの途中から実行することが可能です。
こんな時に便利です。
- ①中断した計算を再開したいとき
他に優先させたい計算ができたときや、計算機のトラブルなどで、計算を中断させてしまった場合でも、正しく計算された部分の続きから計算を再開させることができます。
- ②同じような分子系を評価するとき
分子数が異なるだけの計算の場合、分子の力場設定ディレクトリを残した状態で計算を始めれば、力場設定処理が省略され、指定分子数で溶媒分子配置以降の処理が自動実行されます。
② MD解析
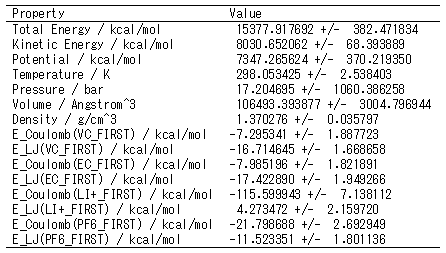
エネルギーや温度などの時間変化や統計値
系全体のエネルギーや温度、密度などの時間変化を見ることができます。溶質分子と溶媒分子との間の相互作用エネルギーの時間変化もわかります。
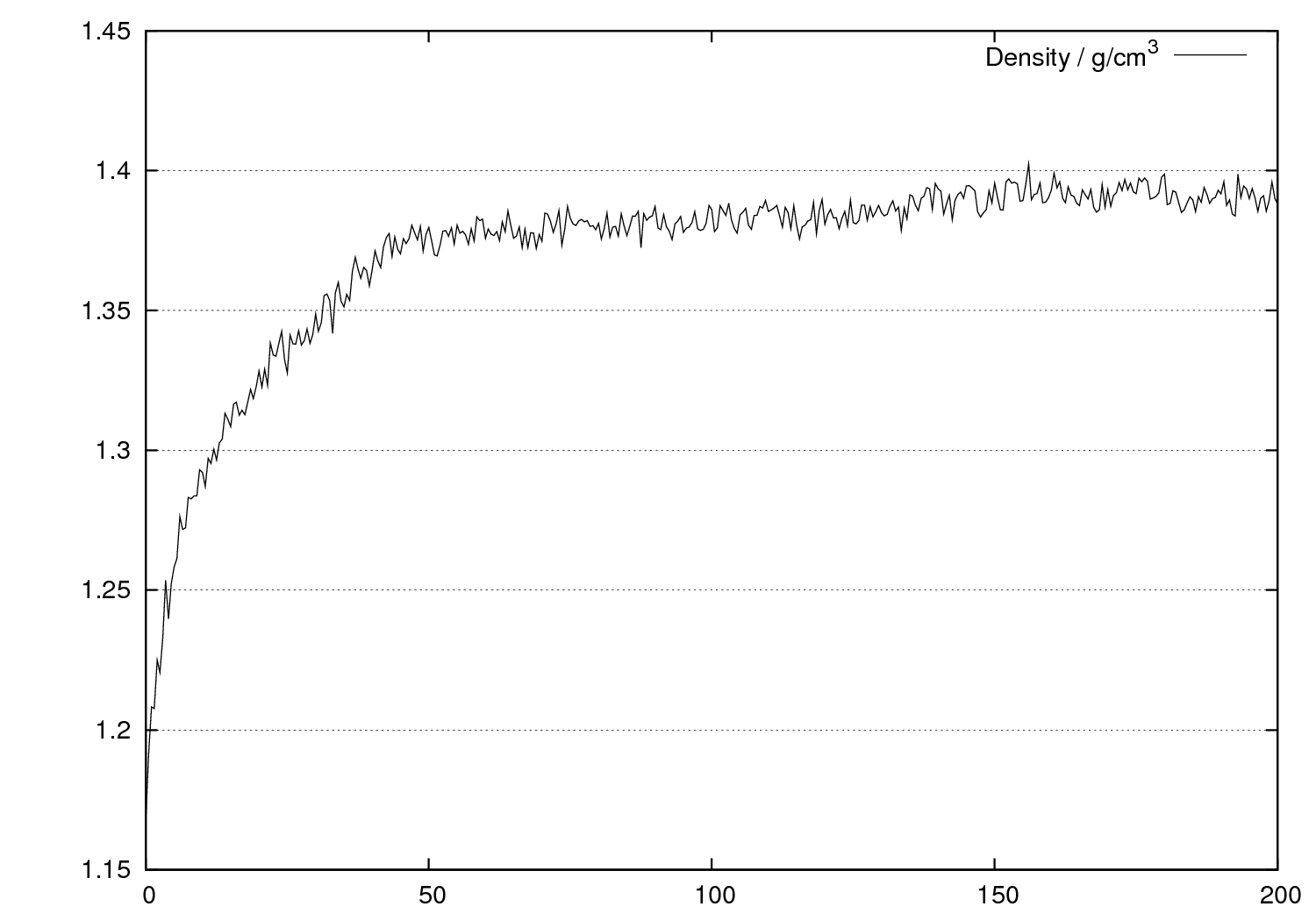
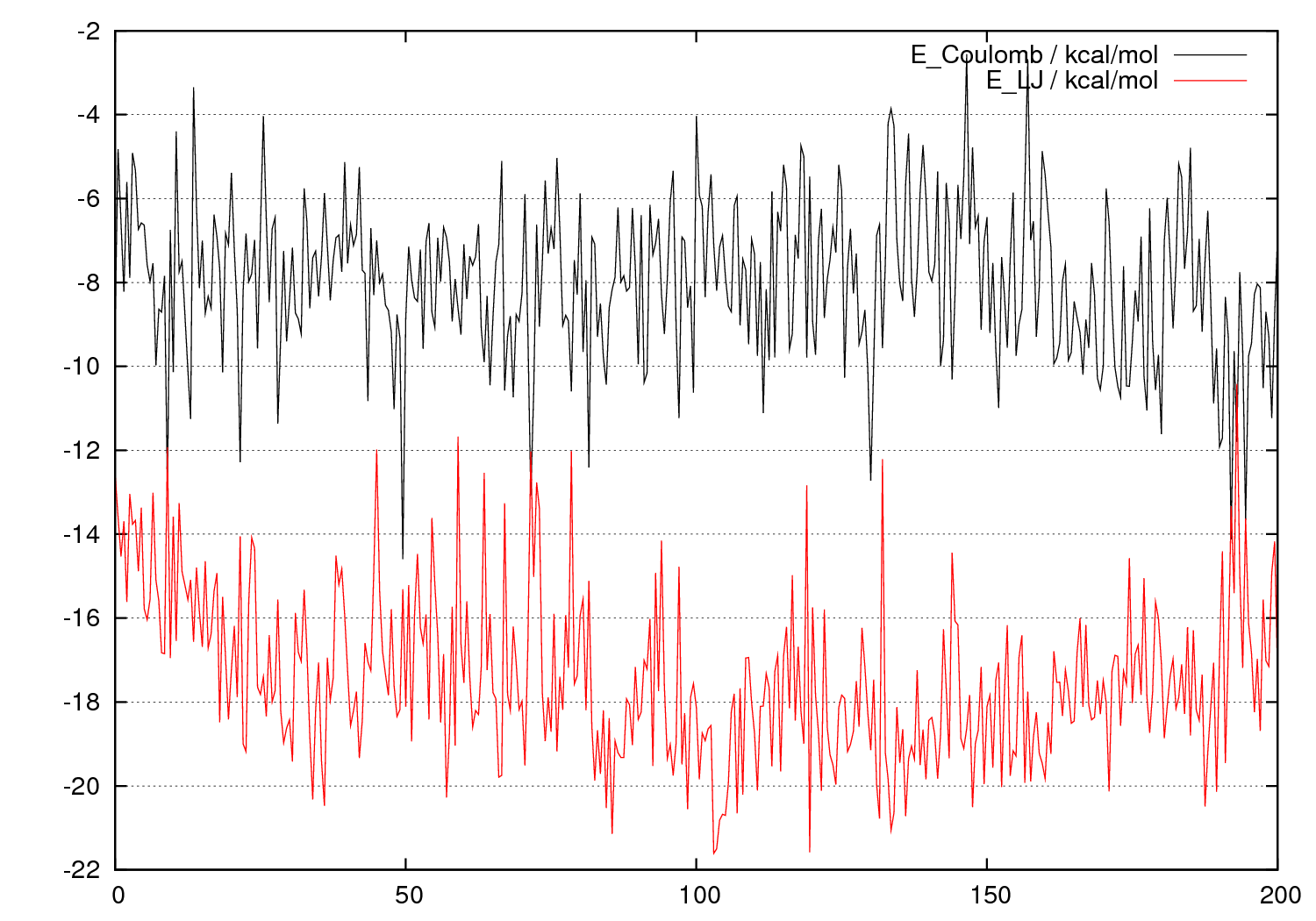
そのほか、統計量(平均値と標準偏差)も出力されます。
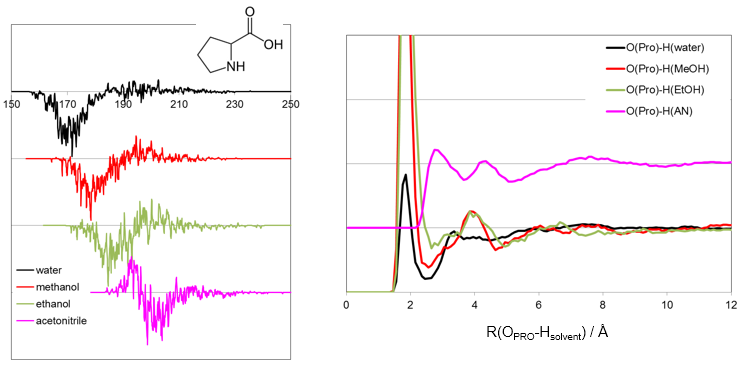
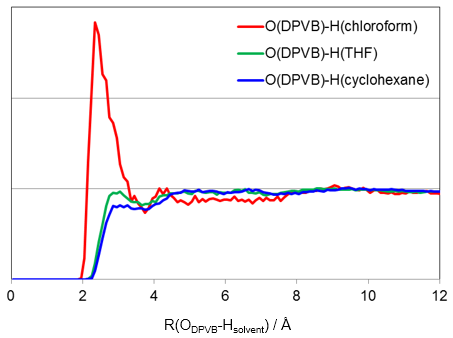
溶媒の分布や拡散の評価
MDのトラジェクトリファイルから溶媒の分布や拡散挙動が解析できます。
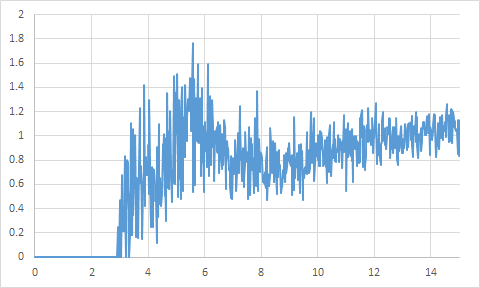
(グラフが粗いのは時間の短いテスト計算のため)
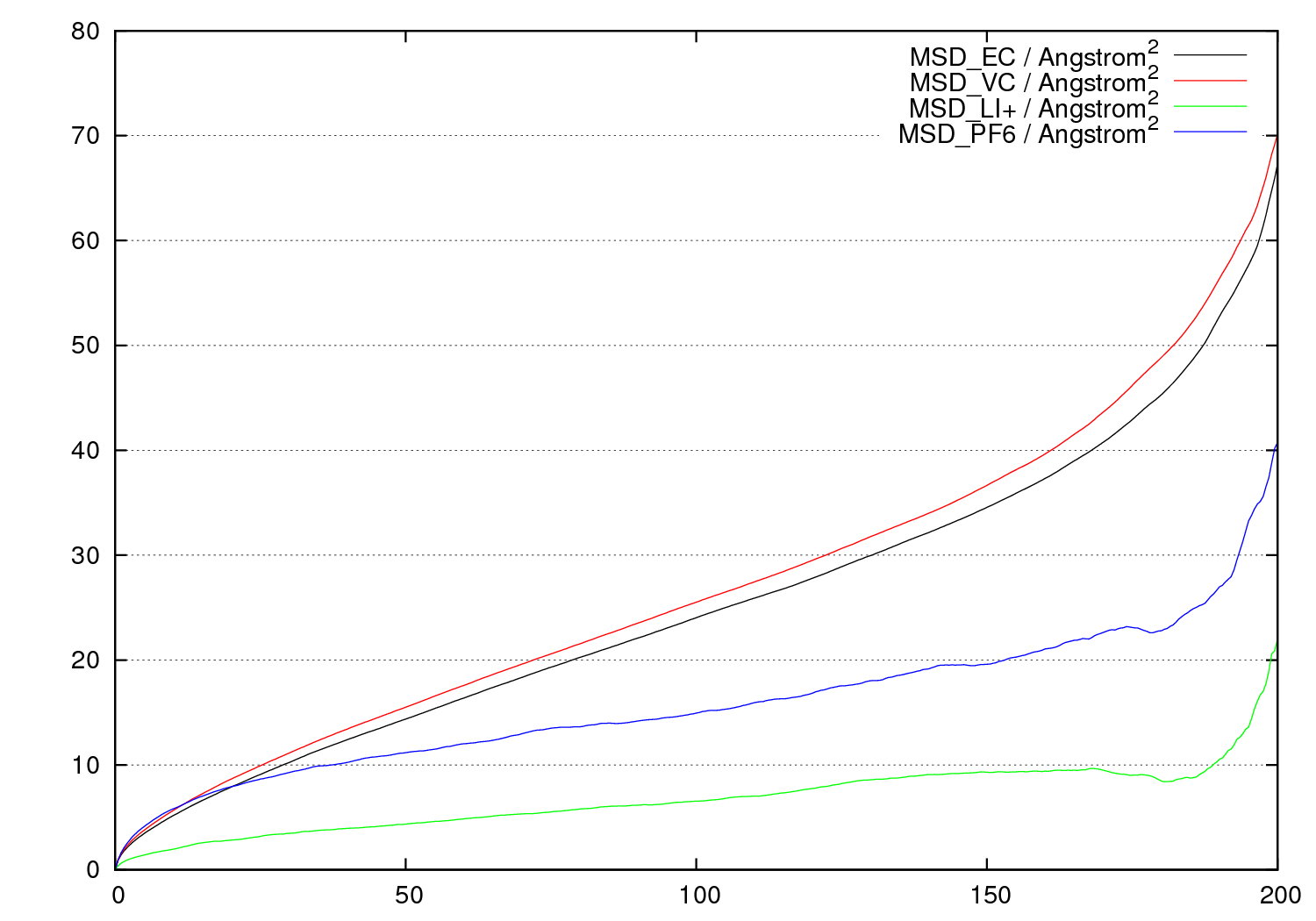
(アインシュタインの関係式より傾きが拡散係数に相当する)
③ 量子化学ツール
一括編集
たくさんのGaussianインプットファイルを一括編集することができます。
例えばこんな編集ができます。
- すべての計算が8並列で実行されるように書き換える。
- すべての計算でchkファイルが出力されるように書き換える。
- すべての分子の計算条件(ルートセクション)を書き換える。
- すべての分子の電荷やスピン多重度の値を書き換える。

一括実行
たくさんのGaussianを一括実行することができます。使用するCPUコア数を制限することもできます。計算終了後は自動的にchkをWindowsやMac環境で読むことのできるfchk形式に変換します。
④ 量子化学解析
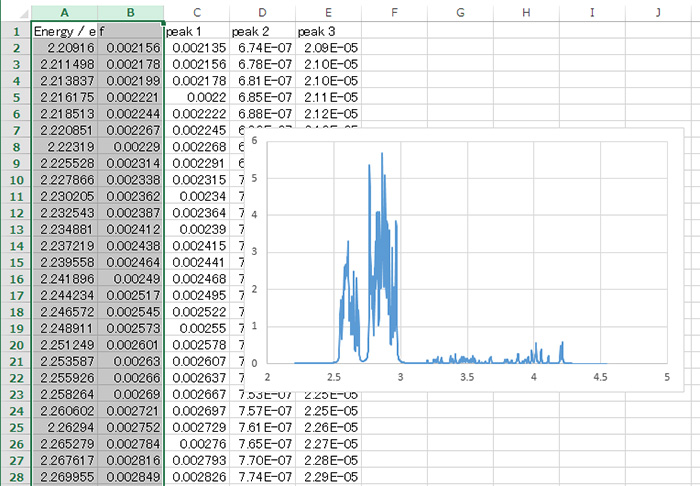
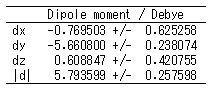
エネルギーや双極子モーメントの統計量
量子化学解析機能では、たくさんのGaussianアウトプットファイル使用した統計解析ができます。
エネルギーや双極子モーメントの平均値のほか標準偏差も計算されますので、分子構造の硬さ・柔らかさなど、新しい視点での現象理解が可能になります。

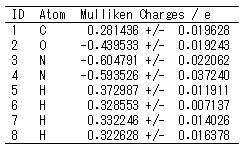
原子電荷
溶媒中で取りうるさまざまな構造に対して評価することで、電子状態の変化しやすい箇所を特定することができます。
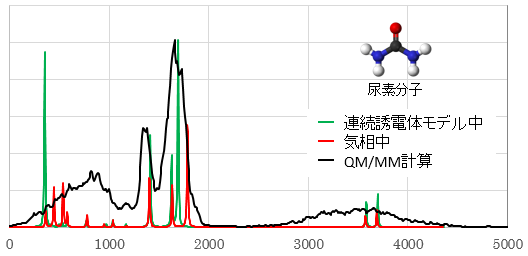
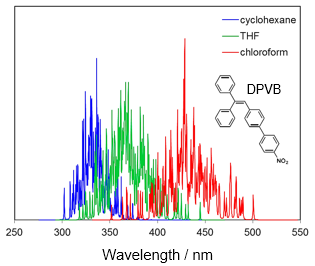
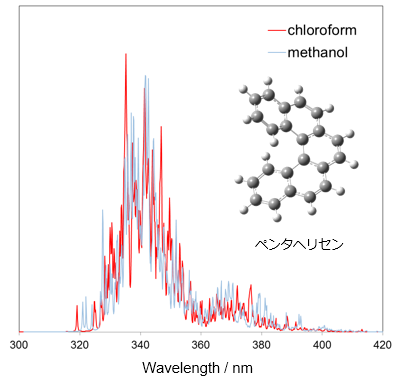
各種スペクトル (IR・Raman・UV-Vis・CD・NMR)
孤立系の量子化学計算では、線スペクトルしか得られませんが、溶液中のさまざまな構造に対する量子化学計算の結果を統計処理することで、実際のスペクトルに対応する幅を持ったスペクトルを評価することができます。
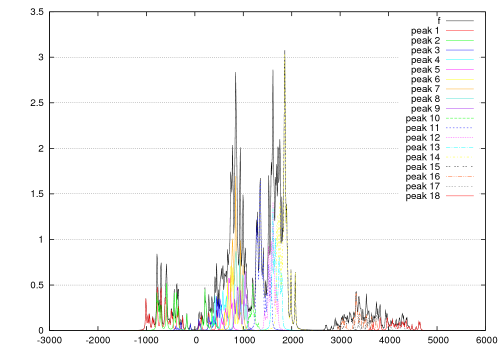
振動成分ごとに色分けされる。
(最適化構造で計算しているわけではないため虚数振動が現れている)
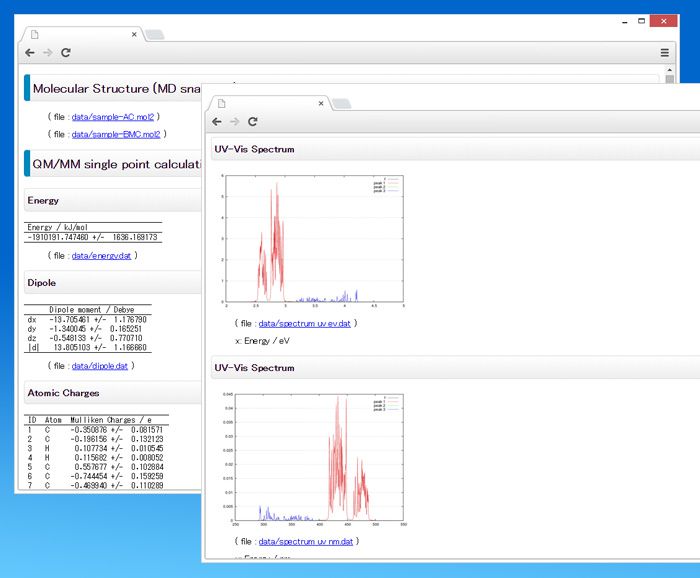
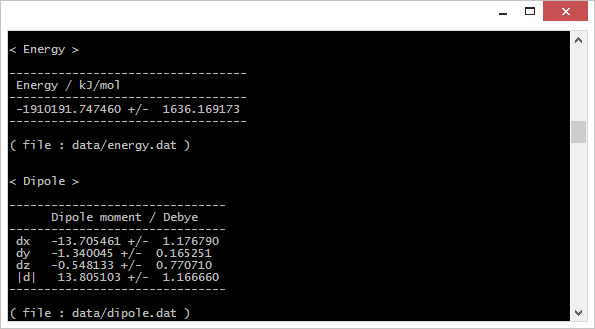
⑤ レポート出力
HTMLレポートとテキストレポート
計算結果はHTMLファイルにまとめられるので、ブラウザで結果を一覧することができます。タブ区切りの数値データファイルは同じ場所のdataディレクトリにまとめて保存されます。
HTMLファイルのほかテキストファイルにも同様の内容が出力されますので、GUIの利用できない環境でも計算結果の確認が可能です。
使い方
自動計算プログラム SOLUTION の使い方を説明します。
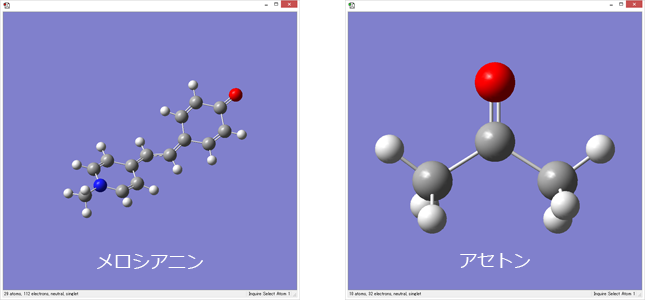
1. 分子を作成します
溶質分子や溶媒分子はGaussViewなどで使い慣れたツールで作成することができます。
2. 計算条件を設定します
分子構造ファイルと分子数を指定し、温度などの計算条件を指定するだけで、設定完了です。
MD計算に詳しい方であれば、MD計算過程の細かい指定も可能です。

3. 計算を実行します
コマンドはたった1行。あとは計算が終わるまで待つだけです。
計算結果はHTML形式で出力されますので、ブラウザで一覧することができます。
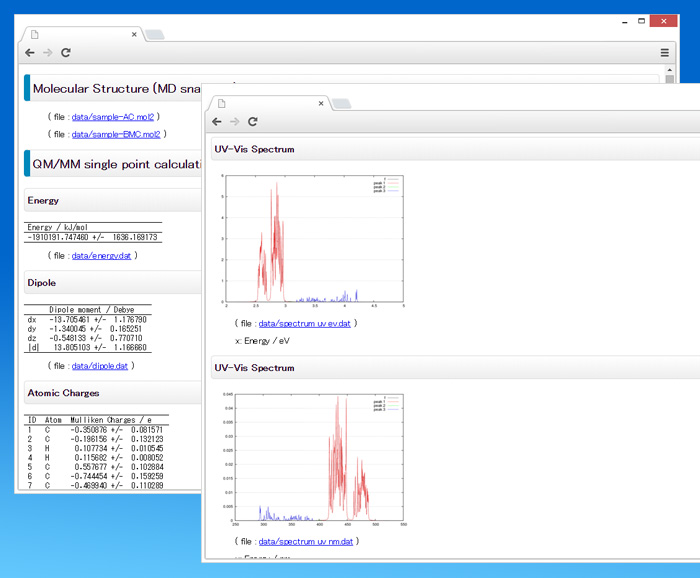
計算データはタブ区切りの数値データファイルとして出力されますので、お使いの表計算ソフトウェアなどを使って表示・加工することができます。
計算例
仕様・価格
動作環境
Red Hat Enterprise Linux 6.x または CentOS 6.x、および、下記ソフトウェア
| ソフトウェア | |
|---|---|
| Python 2.6 | ◎ |
| LSF, LAVA, Grid Engine のいずれか※1 | ○ |
| Gaussian09※2 | ○ |
| AMBER14※3 | ○ |
| AmberTools14 | ◎ |
| GROMACS 4.6または5.0 | ◎ |
| acpype.py | ◎ |
| Open Babel 2.x※4 | ◎ |
| Gnuplot※5 | ○ |
| Image Magick※5 | ○ |
◎=必須 ○=推奨
※1 複数ノードで計算する際に必要になります。プログラム内で呼び出されるAMBER等の外部プログラムがジョブスケジューラによって投入されます。
※2 力場処理(原子電荷)の精度が向上します。また、ab initio QM/MMによるMD計算や解析計算をする場合に必要になります。
※3 古典MD計算が高速化されます。
※4 Open Babel 2.2.3にて動作確認しています。
※5 グラフ画像の自動生成をする際に必要になります。gnuplot 4.2 patchlevel 6, ImageMagick 6.5.4-7 にて動作確認しています。他のバージョンでは正常動作しないことがあります。
構成内容
| 機能 | ツール名 | 説明 |
|---|---|---|
| MD自動計算 | SOLUTION | MD計算や量子化学計算を活用した溶液シミュレーションの実行※1 |
| sander_plus | Sanderの機能拡張版※2 | |
| MD解析 | AMBINFO | AMBERのアウトプットファイルから情報を抽出 |
| GMXINFO | GROMACSのアウトプットファイルから情報を抽出 | |
| TRJ2GJF | AMBERトラジェクトリからGaussianインプットを作成 | |
| 量子化学ツール | GAUEDIT | 複数のGaussianのインプットファイルを一括編集 |
| GAUCUBE | 複数のGaussian cubeファイルから平均分布のcubeファイルを生成 | |
| GAURUN GJOB GJOB-KILL | 複数のGaussianインプットファイルをジョブ投入 | |
| 量子化学解析 | GAUINFO | 複数のGaussianのアウトプットファイルから情報を抽出 |
| レポート | - | 解析結果を成形して出力 (SOLUTION, AMBINFO, GMXINFO, GAUINFOに装備) |
※1 Sander plusをGaussianと組み合わせて使用するためのsander_plus補助プログラム(名古屋大学長岡研究室)が付属します。
※2 Sander (AmberTools14) に任意の量子化学計算ソフトウェアを接続するための機能を追加したものです。AmberTools14のライセンスに基づき、GPL (GNU General Public License) での提供となります。
価格
| QMMM plus |
|---|
| 100万円 / 1ノード ( アカデミック 40万円 ) |
※ 表示価格は税抜価格です。
※ 保証期間は購入後1年間となります。ソフトウェアの活用方法に関するサポートは別途「QMMM plus 使い方サポート」をご利用ください。
サポートオプション
| QMMM plus 使い方サポート | ||
|---|---|---|
| メールサポート | 出張サポート | |
| 内容 | 質問メール対応(ひと月あたり4回) + 使い方セミナー受講(初回のみ) ※1 | 対面での質問対応 ※1 |
| 価格 | 80万円/年 | 25万円/日 (10時~18時) |
※1 遠隔地での実施の場合は別途出張費が発生することがあります。
サポート
► カタログのダウンロード
► よくある質問
よくある質問
ご購入前の質問
- カタログはありますか?
- カタログや計算事例集は、こちらのページからダウンロードできます。
- 体験版はありますか?
- お客様の計算機環境を確認の上、期限付のライセンスを発行させていただいております。詳細につきましては、お問い合わせ下さい。
- リース契約は可能ですか?
- 通常は買い取りにてのご購入となっておりますが、お客様のご都合に合わせて提案することは可能です。お問い合わせください。
- サポートはありますか?
- 有償サポートをご用意しております。詳細はこちらをご覧ください。
ご購入後の質問
- バージョンアップは有償ですか?
- マイナーバージョンアップに関しましては、保証期間内に限り無償でアップデート可能です。アップデート方法につきましては、リリース時にメールにてご案内いたします。
計算の実行・停止に関する問題
- 計算を強制停止する方法が知りたい。
- プログラム本体のpythonから子プロセスのpythonが起動され、その中でジョブスケジューラや各種プログラムが動作しています。以下の手順で停止させてください。
① プログラム本体のpythonをkillする
② プログラム本体から起動しているpythonがあれば、それをkillする
③ ジョブスケジューラに登録されているジョブや実行中の各種プログラムをkillする
- 計算を途中から始めたい。
- QMMM plusでは、ファイルの有無やディレクトリの有無を判断材料にして、次のステップの計算手続きを開始しています。
a) 途中で計算を止めた場合、最後の処理を行っているディレクトリを削除してから、再度計算を始めます。
b) 計算内容が似ているものを計算したい場合、ディレクトリ構成がおなじになるようにコピーすることで、一部の計算を省略することができます。
c) 作成されているファイルを書き換えたのち再度計算を実行することで、プログラム仕様外の計算も可能です。
- 実行後にエラー終了してしまう。
- ジョブ管理ソフトの設定に誤りがあると正常に動作しません。
- 力場生成の段階でエラー終了してしまう。
- a) Gaussianインプットが典型的なフォーマットの場合のみサポートされています。例えば、以下の場合にはエラーになったり、思わぬ計算内容で実行されたりします。
・Z-matrixで書かれているもの
・原子名と座標の間に0が挿入されているもの
・ダミー原子が含まれているもの など
b) 分子の電荷やスピン多重度が不自然の場合にエラーとなります。
c) いくつかの条件が重なると自動処理が正常に働かず、エラーとなることがあります。その際には分子ファイルのファイル形式を変更したり、生成に失敗した力場パラメータファイルを手動で編集したりするなどして対応してください。
- インプットで与えた分子構造と違う構造で計算されてしまう。
- 分子の力場パラメータを生成する際に電荷計算を行いますが、その際にデフォルトでは構造最適化を実行します。構造最適化をしないようにするにはffopt = noを指定してください。
- MD計算開始時にエラーになってしまう。
- a) 分子系のサイズが小さすぎる場合、MD計算時の周期境界条件処理の問題からエラー終了してしまうことがあります。全体の分子数を増やすなどして対応してください。
b) 分子系全体の電荷がゼロになっていない場合、エラーになることがあります。Na+やCl-などイオンを追加指定するなどして対応してください。
AMBERなどのプログラムが正しく設定されていない場合、正常動作しないことがあります。
QM/MM計算の場合、qmconditionにGaussianインプットファイルを指定することができますが、実際の計算とGaussianインプットファイルの設定が不適合の場合にエラーとなります。
- 量子化学計算が行われない。
- calctype = allが指定されていない場合には量子化学計算は実行されません。
MD計算の最後のプロセスをGROMACSで行う場合、量子化学計算は実行されません。
計算終了後の質問
- 双極子モーメントの統計値がおかしい。
- 分子の1スナップショットに対し、双極子モーメントdx,dy,dz,|d|が計算されます。複数のスナップショットについてはこれらの値がそれぞれ独立して統計処理されるため、必ずしも|(<dx>,<dy>,<dz>)| = <|d|>となるとは限りません。
- スペクトルのグラフが粗い。
- a) n_dataの数が少ない場合、グラフは荒くなります。スペクトルの形状にもよりますが、最低でも200はないと傾向が判断できるようなグラフにはなりません。
b) MD計算の最後のプロセスで出力しているトラジェクトリの量が不十分な場合、n_dataに満たない本数の量子化学計算となります。t / dt_outputの値が200以上になるようを調節してください。
- 動径分布や平均二乗変位のグラフが粗い。
- MD計算の最後のプロセスで出力しているトラジェクトリの量が不十分であると、グラフは粗くなります。t / dt_outputの値が10000以上になるよう調節してください。
- 計算結果のファイルにグラフが表示されない。
- a) 動径分布関数(RDF)については原子の組み合わせの数が膨大になるため、グラフは作成されません。
b) グラフの自動作成はgnuplotを用いて行っておりますが、gnuplotのバージョンによっては正しくグラフ生成が行われないことがあります。



お問い合わせ
平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)