サンプリング
いよいよこのチュートリアルの最後のステップ、サンプリングです。何かしらの物理量を計算するステップです。
前回
- 導入
- エネルギー最小化
- NVT平衡化
1 相互作用パラメータ決め
最初のエネルギー最小化で作成した system.top をそのまま使います。
2 初期座標決め
平衡化の処理で得た最後の構造 step2_nvt.gro が初期構造となります。
3 MDの条件決め
以下の内容で sampling.mdp というファイルを作ります。
integrator = md-vv
dt = 0.001
nsteps = 100000
nstenergy = 100
nstxout = 100
coulombtype = PME4 MD実行
まずは読み込みを行います。
gmx grompp -c step2_nvt.gro -p system.top -f sampling.mdp -o step3_sampling.tpr
ここでもNOTEが出てきますが、前回同様に無視します。これで step3_sampling.tpr が生成されましたら、いよいよサンプリングMDを実行します。
gmx mdrun -deffnm step3_sampling性能によりますが、3分~10分程度で終わるかと思います。
5 結果の解釈
動径分布関数
動径分布関数は、原子間の距離を測り、その分布を調べたものになります。これが分かると分子がどれくらい配向しているかなど、さまざまなことが推測できます。
まず、次のコマンドを実行します。
gmx make_ndx -f init.groすると、
here are: 2165 Water residues
0 System : 6495 atoms
1 Water : 6495 atoms
2 SOL : 6495 atoms
nr : group '!': not 'name' nr name 'splitch' nr Enter: list groups
'a': atom '&': and 'del' nr 'splitres' nr 'l': list residues
't': atom type '|': or 'keep' nr 'splitat' nr 'h': help
'r': residue 'res' nr 'chain' char
"name": group 'case': case sensitive 'q': save and quit
'ri': residue index
>という出力が出てくると思います。最後の > はプログラムがこちらの入力を待っている状態を示します。ここで
> a OW
> a HW1 HW2
> qと入力します。すると最後のqで対話モードが終了し、正しく処理されていれば index.ndx というファイルができています。 このコマンドで何をしているかというと、何番目の原子がどのグループに属しているかを設定していて、 index.ndx はその設定を記録したファイルです。たとえば SOL というグループは溶媒に当たる原子が割り当てられています。また、今回の操作では OW グループに酸素原子を、 HW1_HW2 グループに水素原子を割り当てました。このグループはうまく使うと可視化の時に必要な原子団だけを取り出せるほか、MDの時にも特定の原子団だけに熱浴をつけるといったことができます。
とにかくこれで index.ndx ファイルができたのち、
gmx rdf -f step3_sampling.trr -s step3_sampling.tpr -n index.ndx -o rdf_OH.xvg -exclを実行します。すると以下のような出力とともに、どのグループを選ぶかを入力する状態になります。
Available static index groups:
Group 0 "System" (6495 atoms)
Group 1 "Water" (6495 atoms)
Group 2 "SOL" (6495 atoms)
Group 3 "OW" (2165 atoms)
Group 4 "HW1_HW2" (4330 atoms)
Specify a selection for option 'ref'
(Reference selection for RDF computation):
(one per line, <enter> for status/groups, 'help' for help)
>ここでは酸素原子と水素原子をを選びましょう。
> OW
> HW1_HW2(あるいは、単に対応するグループ番号の 3 と 4 を入力しても大丈夫です。)
このあと Ctrlを押しながらDキーを押しますと、軌跡ファイルの読み込みをして動径分布関数の計算に入ります。なお、テスト環境では実際のMDより時間がかかりました。計算が終わると rdf_OH.xvg というファイルができているかと思います。これを可視化したところ、下図のようなグラフが見えるかと思います。
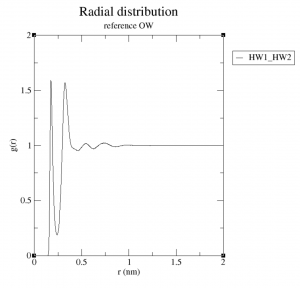
これは酸素原子からみたときの水素原子の分布です。 0.2 nm 付近のピークが鋭く大きいのは、強い静電力により水素原子が酸素原子近くに束縛されているためと思われます。また0.4 nm、0.6 nm、・・・とピークが並びますが、だんだん弱く広くなっています。 最短位置のピークの高さ・幅、ピーク減衰の比率などは系によりさまざまですが、おおむね液体ではこのように遠くに行くに従い構造が乱れていく傾向が見られます。
軌跡の可視化
軌跡のデータは step3_sampling.trr にあります。 少々重いですので、コピーなどする際には気を付けましょう。 これと最初の座標データ step2_nvt.gro があれば、VMD を使って分子の運動を動画で表示できます。
- VMDを立ち上げます。
step2_nvt.groを読み込みます。- VMD Main窓のリストにて、右クリックなどで「Load Data Into Molecule…」を選択します。
- Filenameで
step3_sampling.trrを選びます。また、file typeで「Gromacs TRR Trajectory」を選びます。 - 「Load」ボタンをクリックします。読み込みが終わったらMolecule File Browserを閉じます。
- 周期境界条件で結合が横断すると見栄えが悪いです。この時は「Extensions」→「Tk Console」を開き、
pbc unwrapコマンドを実行します。
すると、水分子が徐々に拡散していく様子が見えるかと思います。
2番目のstep2_nvt.groを読み込んだ直後。
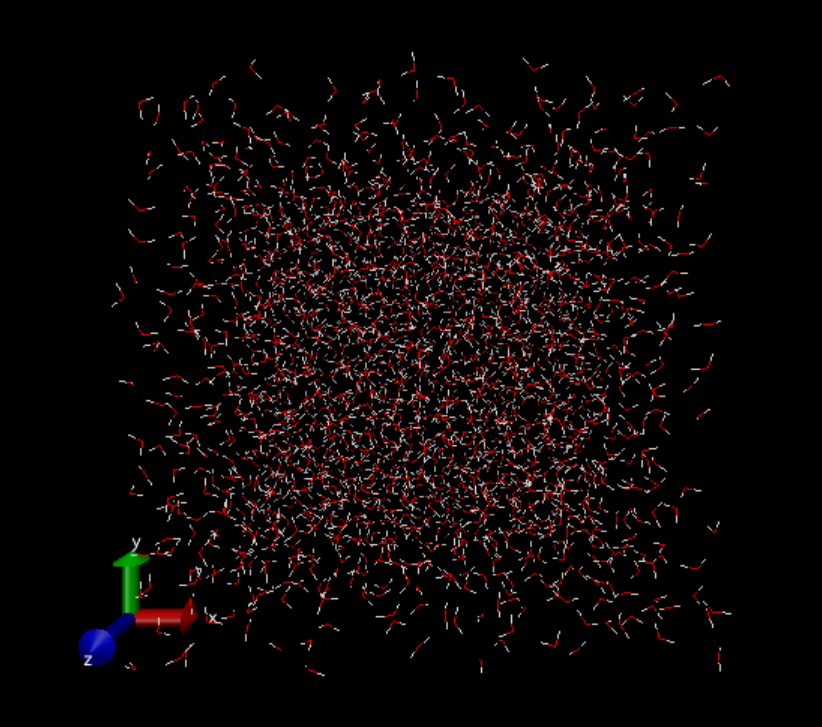
5番目で全フレームを読み込んだあと。
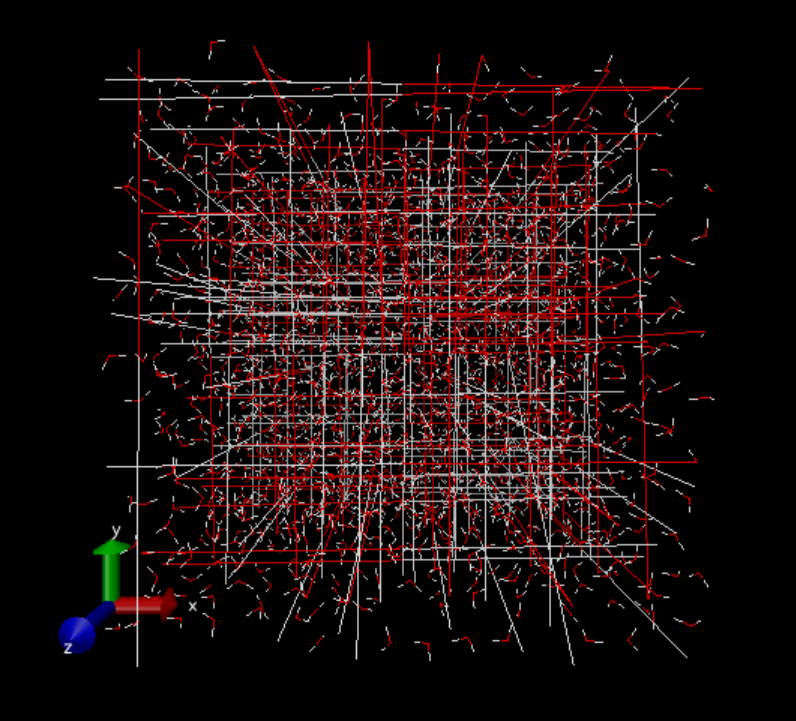
6番目のpbc unwrapを実行した後。 箱を超えて水分子が広がっているように見えます。
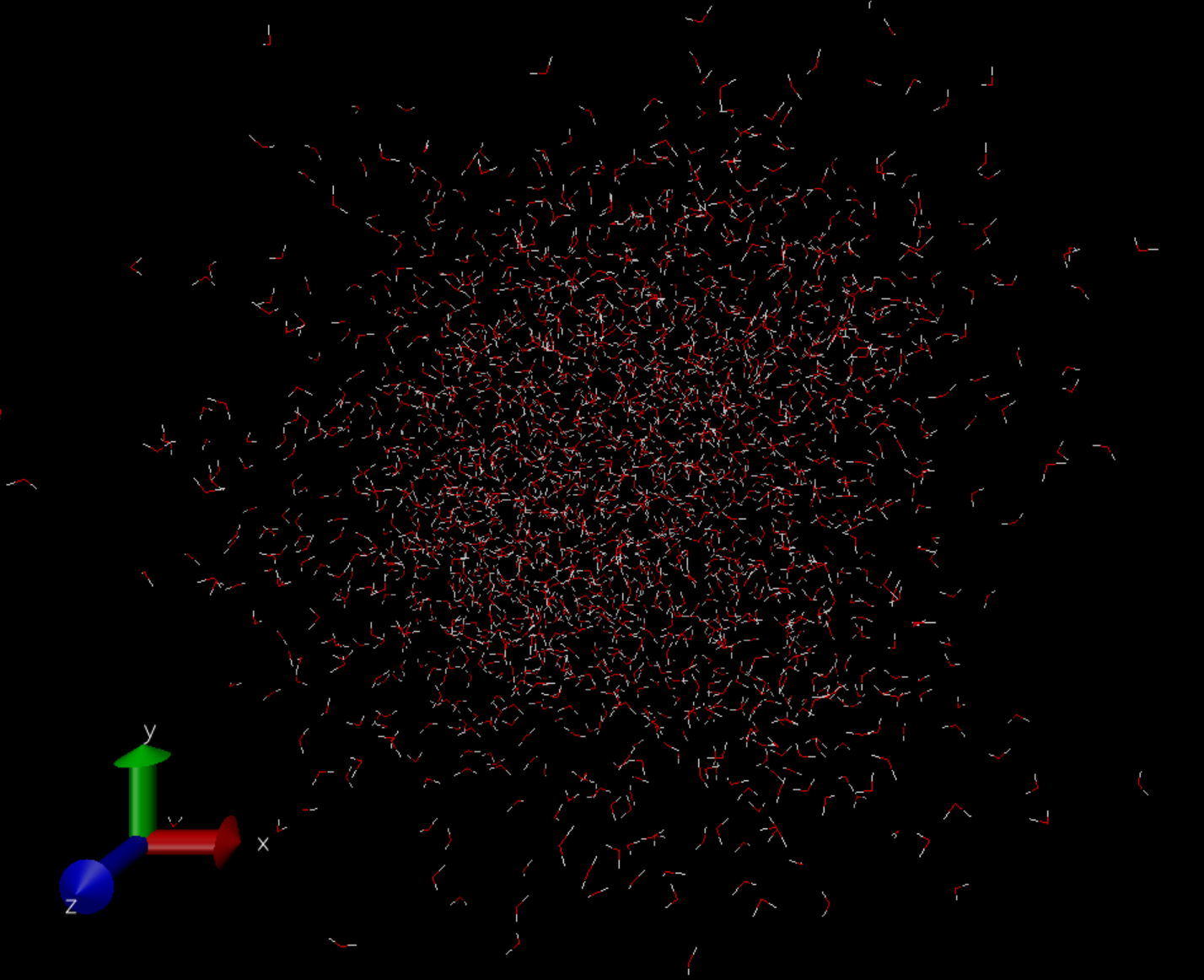
補足しておきますが、これは真空中に置かれた数千個の水分子のMDではありません。実際の計算では座標は周期境界長さで折りたたまれ、そして分子がコピーされたイメージセルが縦横に無限に広がっている状態となります。密度はちゃんと保たれ、凝縮相のMDが行われています(でないと動径分布関数がちゃんと出ません)。可視化の都合上で真空中を広がっていくように見えています。
ここには載せきれませんでしたが、VMDでしたら実際に水分子が動くアニメーションが見られるはずです。
ただ可視化の絵にも不満点が多いかもしれません。この簡単な水分子のMDだけでも、筆者がこだわる場合は以下の点を直したいと思っています。
- 分子を球体ー棒モデルで表示したい
- 周期境界を超えた分子は遠くに行くのではなく、反対側からの壁から出てきてほしい
可視化もMD自体に負けず劣らず様々なテクニックやノウハウが要求されます。
6 まとめ
これでチュートリアルとして考えている作業は一通りやったことになります。
しかし実際の問題をMDで解釈しようとすると、
- 水溶液中の分子・高分子を見たい
- 水以外の溶媒を試したい
- 圧力をコントロールしたい
- 界面を見たい
- 不均一な系を見たい
- 人工的に力を加えたい
- 物理的にどれくらい妥当か判断したい
といった欲求が出てくるかと思います。今後はこれらの一端に触れられるようなチュートリアルを用意できたらと思います。
