エネルギー最小化
ここから実際にファイル作成やコマンドを実行し、MDを行っていきます。
前回
- 導入
最初の準備
今回は以下のファイルをvimやemacsなど、Linux上のテキストエディタで作成します。
- system.top
- em.mdp
- nvt.mdp
- sampling.mdp
また、GROMACSの実行により多くのファイルが生成されます。このため、実行するディレクトリ(フォルダ)は専用のものにしておいたほうがいいでしょう。
ということで、以下のようにディレクトリを作るコマンドを実行します。(なんらかの理由ですでにwater_gromacs_tutorial ディレクトリがあったら適宜変更してください。)
mkdir water_gromacs_tutorial
cd water_gromacs_tutorial以下、ファイル作成などはこの `water_gromacs_tutorial` の中で行うものとします。
無理のない構造の生成
ここでは、水分子を並べたのち、エネルギー最小化という処理を行います。
1 相互作用パラメータ決め
まず、以下のような内容の system.top ファイルを作成します。
#include "amber03.ff/forcefield.itp"
#include "amber03.ff/spce.itp"
[ system ]
water in water
[ molecules ]
最初の2行でどの力場を使うかを指定していて、この場合はAmber03を使うということになります。
今回はGROMACSのコマンドの流れを確認するのが目的のチュートリアルですので、この内容については詳しくは触れません。
2 初期座標決め
水分子を適当に並べ、初期構造とします。
gmx solvate -cs spc216.gro -o init.gro -box 4.0 -p system.top
これで 4 nm 立方の箱の中に水分子を並べることができました。init.groというファイルが新しくできていることがわかりますが、これがすべての水分子の座標を記録しているファイルです。
ここで system.top の中身をみてみると、最終行に
SOL 2138が追加されるはずです。(最後の `2138` は乱数の具合により数%程度前後します。)
ここで、 init.gro ファイルにランダムに配置された水分子の座標が記録されているといいましたが、これはAvogadroなどのソフトウェアで可視化することができます。

3 MDの条件決め
以下のような内容で em.mdp というファイルを作成します。
integrator = steep
emtol = 1000.0
emstep = 0.01
nsteps = 50000
coulombtype = PME今回は物理的な話はしませんので説明は省略しますが、「エネルギー最小化」を行う設定です。(em.mdp の `em` は Energy Minimization の頭文字です。)
4 MD実行
設定ファイルの「コンパイル」
以上の段階で「初期構造」「力場パラメータ」「MD条件」の3つのファイルを作ったわけですが、これらからMDを行うプログラムが読み込めるような形式のバイナリファイルを作ります。
gmx grompp -c init.gro -p system.top -f em.mdp -o step1_em.tpr
こちらのコマンドで mdout.mdp と step1_em.tpr の2つのファイルが生成されたかと思います。
MD実行
ここまできたらあとは実行するだけです。
gmx mdrun -deffnm step1_em
エラーなく終われば、以下のファイルが生成されているはずです。
step1_em.edrstep1_em.logstep1_em.trrstep1_em.grostep***.pdb(step16c.pdbなど。ない場合もある)
このうち step1_em.groがエネルギー最小化の結果の構造を記録したファイルで、次のMDで使うものになります。
補足 オプションについて
-deffnm step1_em は入出力ファイルのファイル名を step1_em に固定するオプションです。ない場合とある場合の対応は以下のようになります。
| オプションなし | -deffnm hoge |
|
|---|---|---|
| 入力 | topol.tpr |
hoge.tpr |
| 実行ログ | md.log |
hoge.log |
| エネルギーのログ | ener.edr |
hoge.edr |
| トラジェクトリ | traj.trr |
hoge.trr |
| 最後の座標 | confout.gro |
hoge.gro |
単発のMDだとオプションが増えて面倒なだけですが、同じディレクトリで何回もMDを行う場合はファイル名が別になって管理が楽になります。
補足 GPU使用時のエラー
次のようなエラーが出て止まることがあります。
Fatal error:
When using GPUs, setting the number of OpenMP threads without specifying the
number of ranks can lead to conflicting demands. Please specify the number of
thread-MPI ranks as well (option -ntmpi).このときは、インストールしたマシンのGPUの台数が n であれば -ntmpi n というオプションをつけて実行すると動くことがあります。たとえば
gmx mdrun -deffnm step1_em -ntmpi 1とします。
5 結果の解釈
実際にエネルギーはどの程度小さくなっているでしょうか。次のコマンドで必要な情報を取り出せます。
gmx energy -f step1_em.edrすると以下のような表が出てきます。
Select the terms you want from the following list by
selecting either (part of) the name or the number or a combination.
End your selection with an empty line or a zero.
-------------------------------------------------------------------
1 LJ-(SR) 2 Coulomb-(SR) 3 Coul.-recip. 4 Potential
5 Pressure 6 Vir-XX 7 Vir-XY 8 Vir-XZ
9 Vir-YX 10 Vir-YY 11 Vir-YZ 12 Vir-ZX
13 Vir-ZY 14 Vir-ZZ 15 Pres-XX 16 Pres-XY
17 Pres-XZ 18 Pres-YX 19 Pres-YY 20 Pres-YZ
21 Pres-ZX 22 Pres-ZY 23 Pres-ZZ 24 #Surf*SurfTen
25 T-rest
今回はエネルギーの経緯が見たいだけなので、
4 0と入力して Enter を押します(人によっては Return かも)。 すると、 energy.xvg というファイルが生成されています。このxvg形式のファイルは、Graceというプログラムで可視化することができます。
詳しくはGraceの使い方の話になるので当記事では省略しますが、Linux上ではGraceがインストールされていれば
xmgrace energy.xvgというコマンドを実行しますと以下のグラフが表示されます。

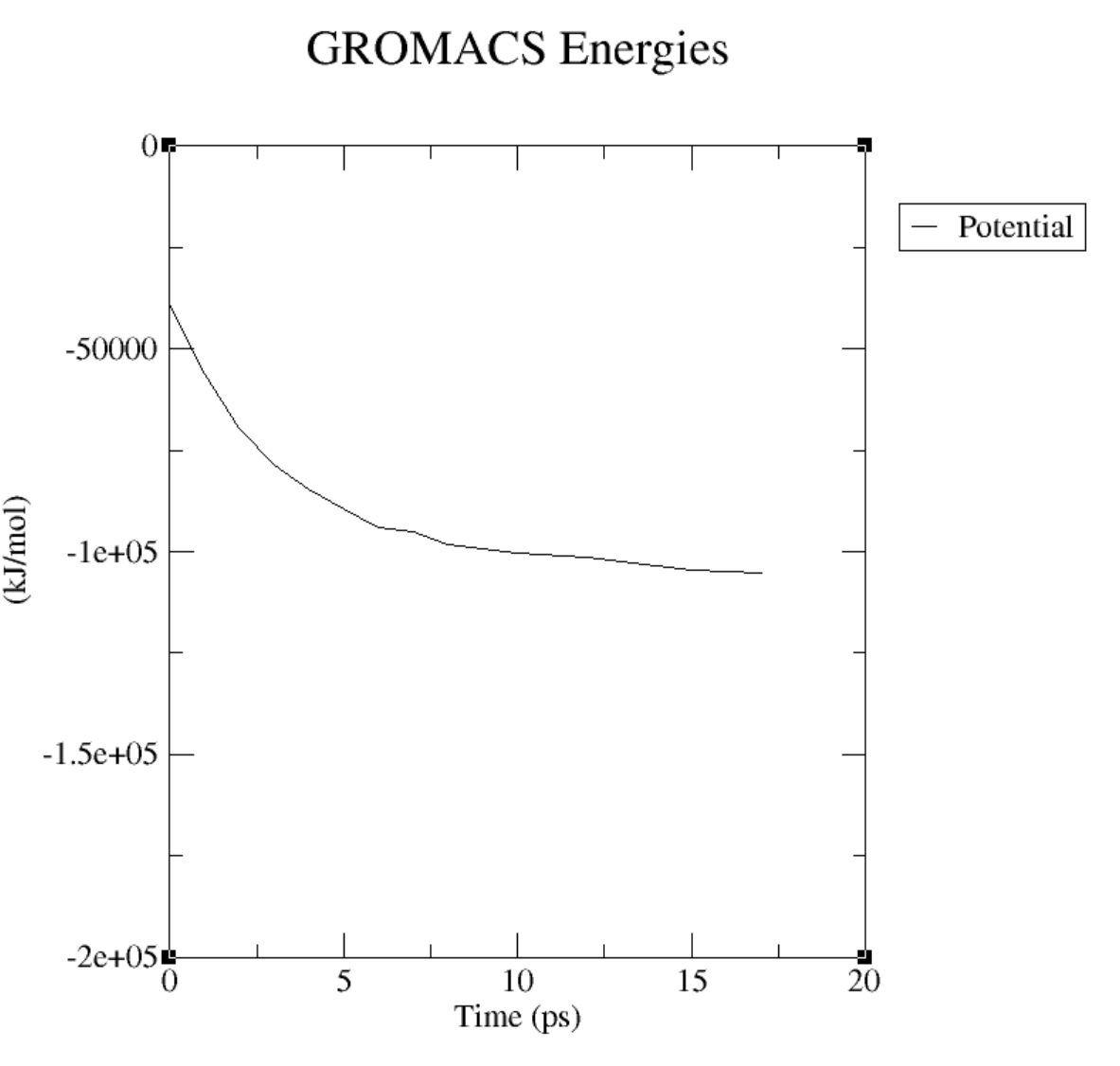
実際にエネルギーが右肩下がりで、ちゃんと最適化されていることがわかります。
気が済んだらウィンドウ右上の[x]ボタンを押して終了します。
6 次のステップへ
エネルギーがちゃんと小さくなっていって、また最後の構造も確認したところ確認したところで、次は実際に温度を与えて動かしてみます。
—次回に続く