





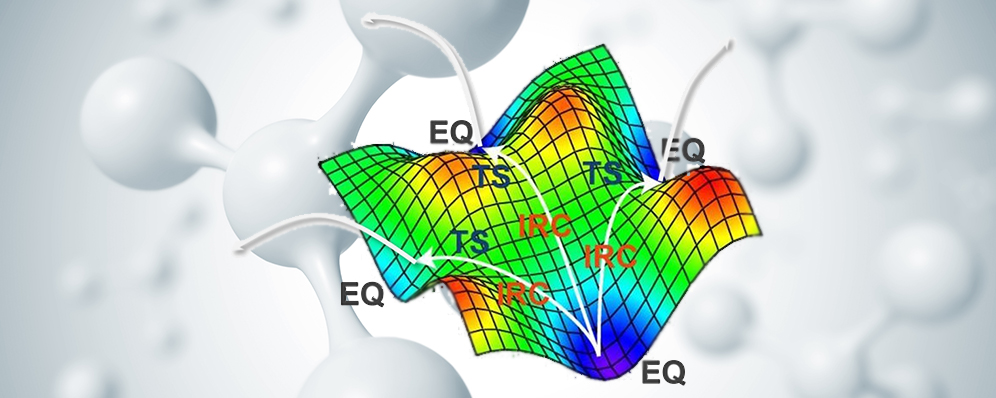
たった1つの分子をインプットするだけで
すべての反応経路を自動探索
GRRMシリーズの最新版GRRM26は、新たに高速エネルギー計算プログラ... すべての反応経路を自動探索
対象の製品はありませんでした。





















平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)