所望の物性を有する分子構造の探索 - データベースレスで実現するMIを用い 新たな新素材開発のステージへ ―
実験・計算・データサイエンスを1つのツールでカバー
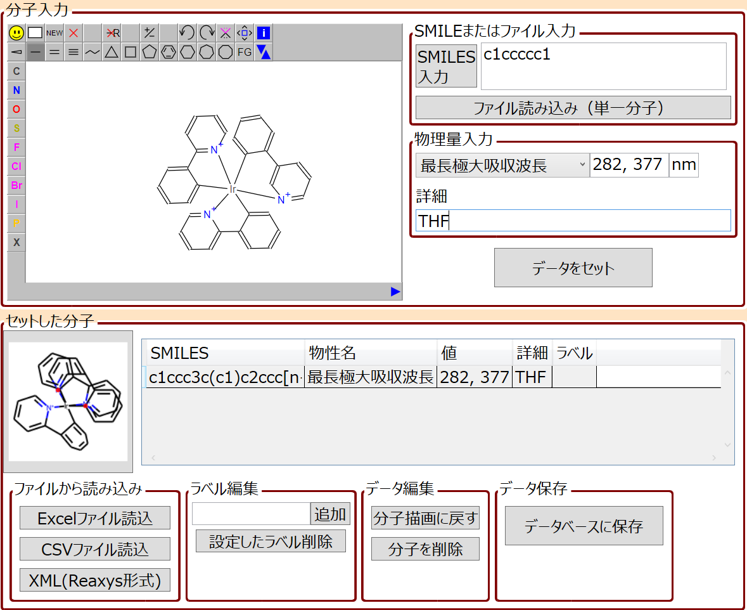
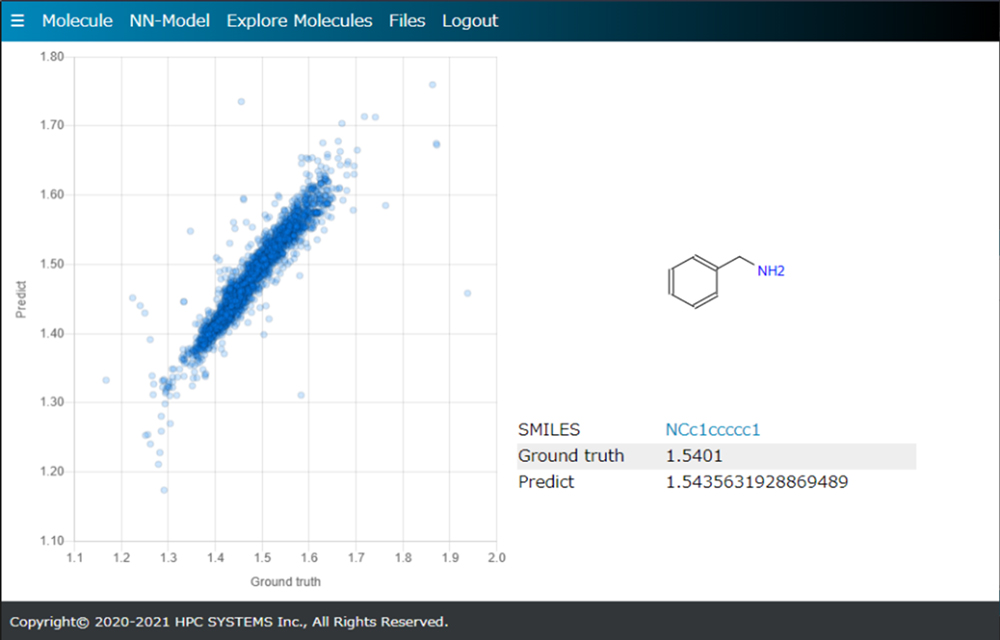
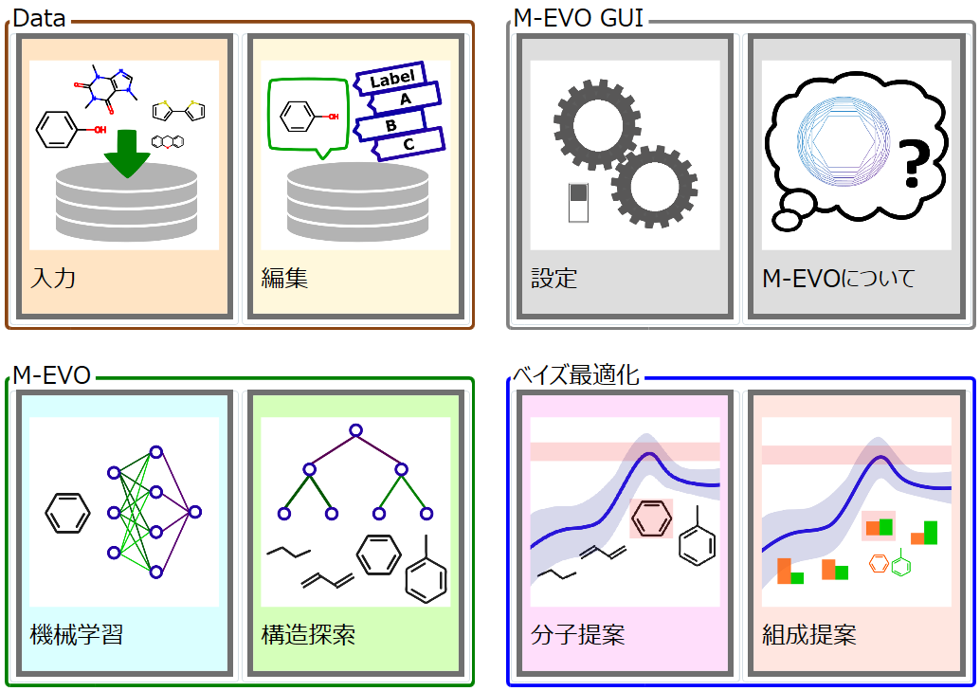
M-EVO®は、開発実験担当者が一人で実験・計算・データサイエンスの3役をこなして、研究開発を効率的に行うためのツールです。
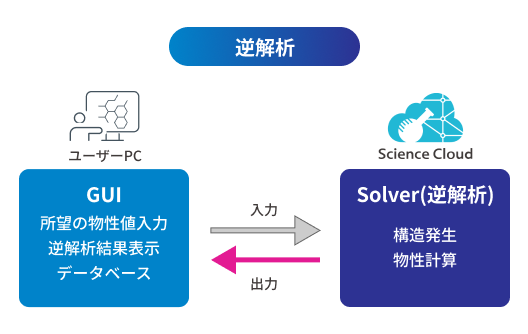
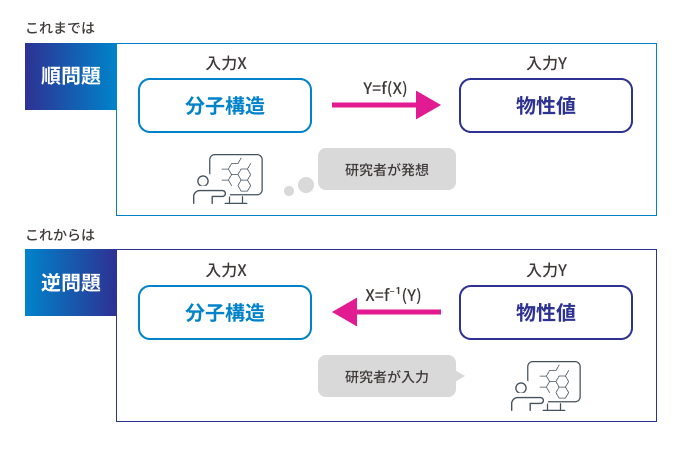
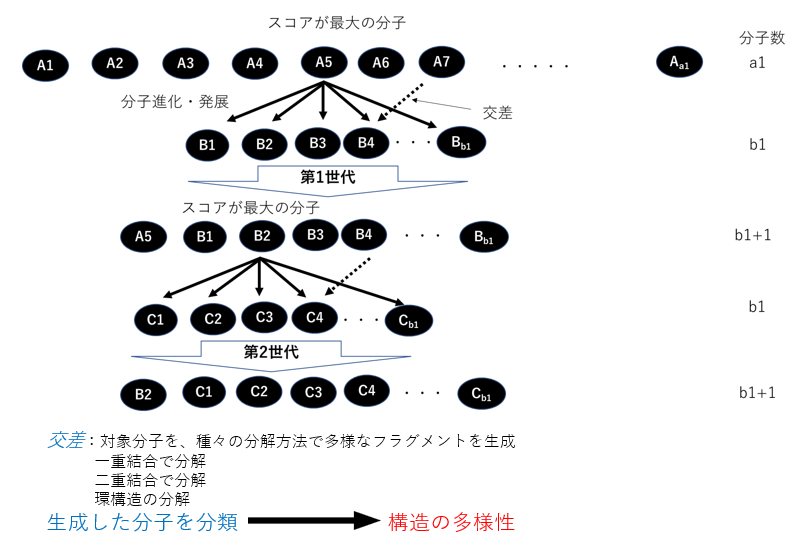
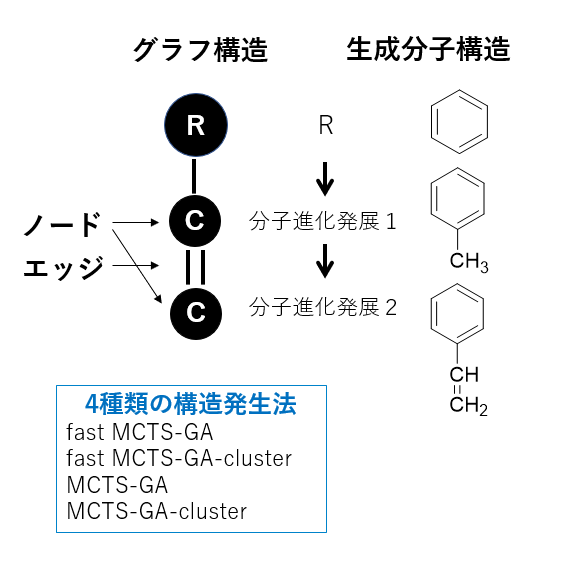
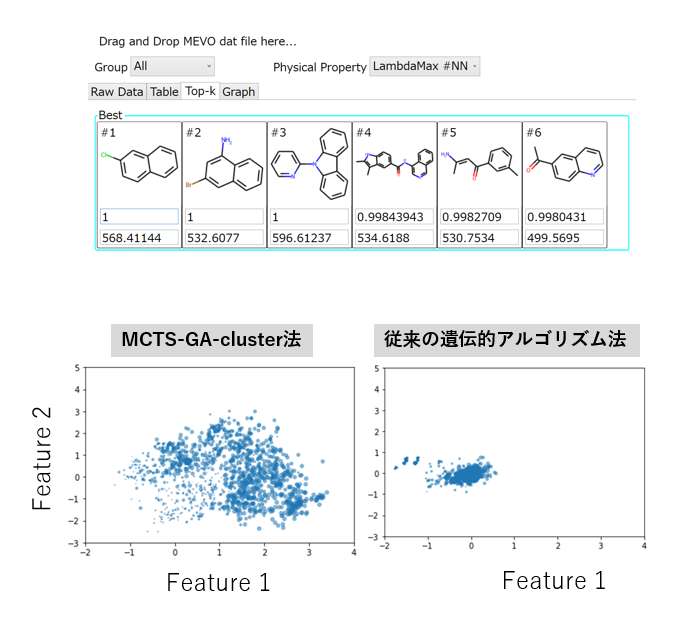
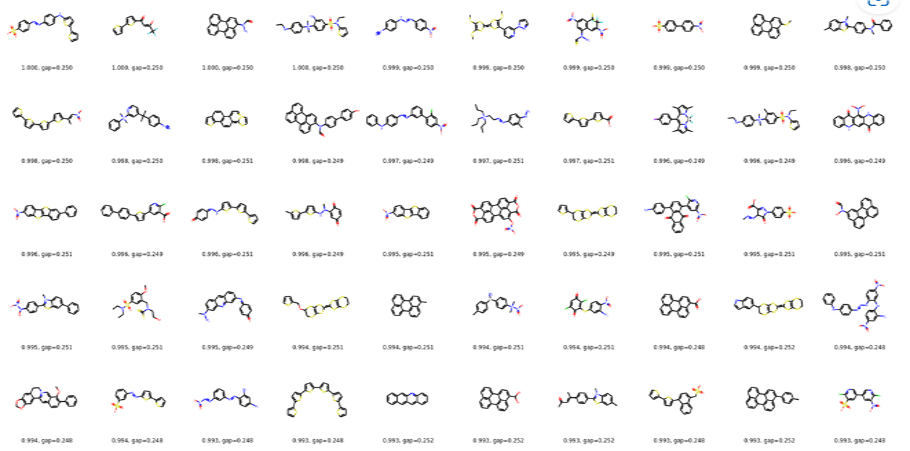
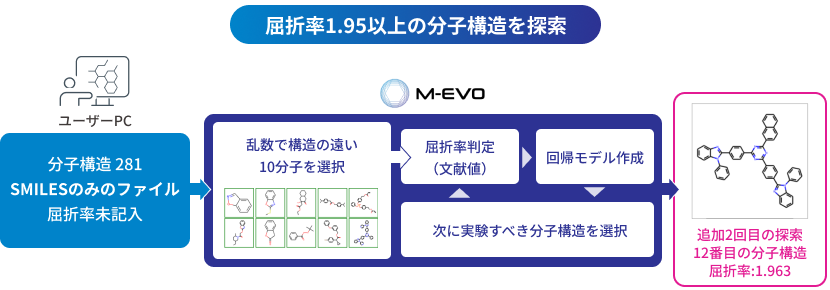
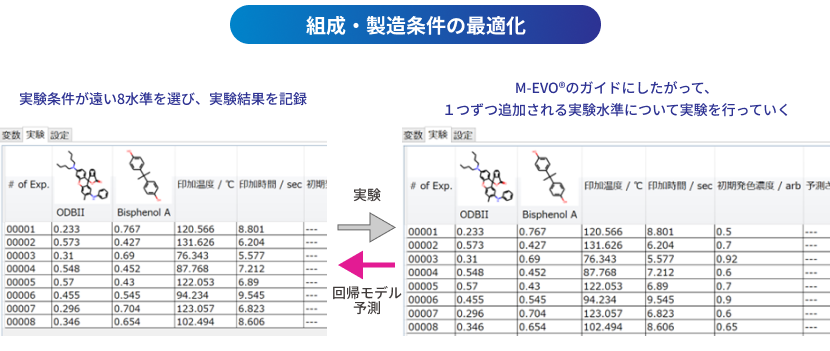
M-EVO®は、当社オリジナルのアルゴリズムにより所望の物性を有する多様な分子構造を探索し、提案します。複数の目的物性を考慮でき、合成の可能性や溶剤溶解性等の指標も含めたスコアを算出し、実用的な分子構造の探索ができます。
M-EVO®による開発フローイメージ
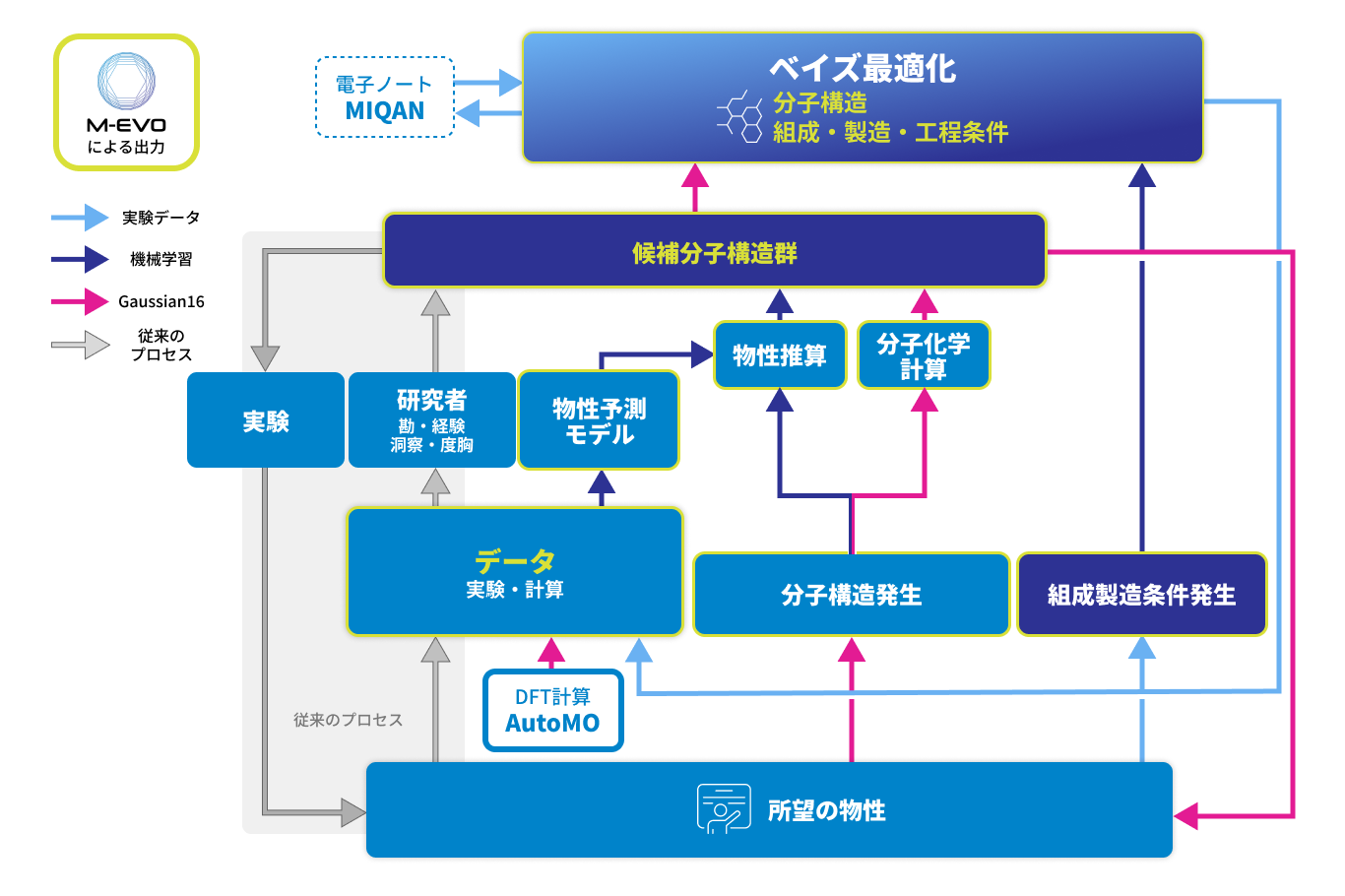
MIを素材の研究開発で有効活用するためには、開発実験担当者と計算担当者とデータサイエンティストの3者が協力することが大切です。現実的には、この3者が同じ土俵で議論することは容易なことではありません。開発担当者は常に実験の様子を見ながらどうしようか考えています。計算担当者は理想的な系で厳密な議論をしがちです。データサイエンティストは多量のしかも均質なデータを要求しがちです。この3者のベクトルをタイムリーに合わせるという難事を、M-EVOを使えば実現可能です。