
Software
計算化学 ソフトウェア
Amber
Amberは米カリフォルニア大学のP. A. Kollmanらのグループにより開発された、生体分子の分子動力学(MD)計算のための力場群であり、またこれらの力場をシミュレーションするためのMDプログラム群です。現在は、米ラトガース大学のD. A. Caseらによるグループにより維持・管理されています。プログラム群は、入力準備プログラム・シミュレーションプログラム・結果解析プログラムに大別されます。
Status
https://ambermd.org/AmberPatches.php
pmemd26 Updates (April, 2026)
update.1: Disable Xray refinement in pmemd; see Section 32.3 of the Amber 2026 Reference Manual for more information.
update.2: Update definitions to allow pmemd to build on AMD GPUs
pmemd24 Updates (April, 2025)
update.1: Support for pmemd.cuda on Blackwell GPUs
update.2: Add plumed to 3rdPartyTools, to facilitate integration with pmemd.
update.3: Updates to support NTP=1, barostat=1 runs for pmemd.cuda on Blackwell GPUs
Amber24 Updates (April, 2024)
update.1: Fix some cases where HREMD could hang with pmemd (parallel CPU version)
update.2: Adds compilation target pmemd.decomp which supports Thermodynamic Integration Atomic Decomposition
update.3: Fixes softcore potentials in pmemd.cuda when gti_add_sc=25
Amber22 Updates (April, 2022)
update.1: Fix two (rare) problems, with 12-6-4 potentials and duplicate TI
update.2: Fix to the default values of soft-core potential parameters scalpha and scbeta.
update.3: Fix the tishake2vite test outputs, so that the test cases now pass.
update.4: Fix a memory leak, speed up NMR calculations in pmemd.cuda.
update.5: Updates scaling in the xray module of pmemd.cuda.
ライセンス
- Amber はバージョンごとに個別のライセンス締結となっています。
- Amber26およびAmber24 では Academic / non-profit / government の方が無料で利用可能となりました。詳細は How to obtain Amber26 のページを参照ください。
特長
溶媒理論3D-RISMとハミルトニアンレプリカ交換法が実装されたことで、計算の精密さが増し、かつより大規模な系での計算が可能となりました。また、構造立脚型の創薬に用いられています。細胞内の環境を再現し、結合自由エネルギーを定量的に求めることができます。GAUSSIANとの連携も可能です。
Amberの基本機能
- 自由エネルギー摂動法や熱力学的積分法などによる自由エネルギー計算
- 溶媒中での自由エネルギー計算
- エネルギー極小化計算
- MDシミュレーション
- 原子間距離や角度の算出
- 温度因子(B-factor)の算出
- 構造変化や構造の揺らぎをみる根平均2乗偏差(RMSd)の算出
関連情報
- Amber入門
- Amber応用例
- Amber ユーザーマニュアル
- QMMM plus 2 – Amberをもっと便利にするための便利ツール (弊社製品、販売終了)
- Amber26の主な新機能
- HPCシステムズがビルドしたAmberの特徴
- 参考書籍:Essentials of computational chemistry: theories and models (著書:CJ Cramer) (外部リンク)
- 参考書籍:コンピュータ・シミュレーションの基礎 (著者:岡崎 進・吉井 範行) (外部リンク)
- 参考書籍:Amberによる生体高分子シミュレーション入門 (著者:Amber研究会) (外部リンク)
- 参考書籍:すぐできる分子シミュレーションビギナーズマニュアル (著者:長岡 正隆) (外部リンク)
- HPCシステムズの計算化学ソリューション – コンサルティング・受託計算・セミナー
ベンチマーク
NVIDIA HPC アプリケーション性能
随時更新されている NVIDIA HPC Application Performance のページを参照ください。
NVIDIA A100 による Amber ベンチマーク結果
NVIDIA_Life and Material Sciences Application Performance Guide
その他
- NVIDIA V100 による AMBER16 ベンチマーク結果 (2017年10月:NVIDIA社提供資料) (PDF:352KB)
- NVIDIA P100 による AMBER16 ベンチマーク結果 (2017年2月:NVIDIA社提供資料) (PDF:360KB)
- NVIDIA K40・K80 Amber14 ベンチマーク比較 (2015年7月10日)
- NVIDIA K80 ベンチマーク結果 (2015年2月17日)
- インテル® Xeon® プロセッサー E7-8800 v2 ファミリー ベンチマーク結果 (2015年2月13日)
- インテル® Xeon® プロセッサー E5-2600 v3 ファミリー ベンチマーク結果 (2014年9月9日)
- インテル® Xeon® プロセッサー E5-2600 v2 ファミリー ベンチマーク結果 (2013年9月11日)
- NVIDIA K10でのAmber 12 GPUベンチマーク結果 (2013年1月7日)
- インテル® Xeon® E5-2600シリーズ 8ノードクラスターでのAmber 12ベンチマーク (2012年10月5日)
- インテル® Xeon® E5-2670 ベンチマーク結果 (2012年3月7日)
- CUDAでAmberを高速化! (2009年)
Amber26 の主な新機能
Improvements to Advanced Methods
- Enhancements to alchemical transformation pathways/softcore potentials and alchemical enhanced sampling (ACES) method:
- advanced lambda scheduling options including new tools for optimized phase space overlap lambda-spacing
- scaffold-hopping and absolute binding free energy capability through lambda-dependent “Boresch” bond, angle, and torsion restraints
- lambda-dependent RMSD-fitting restraints to floating reference molecular scaffolds
- non-equilibrium work framework (Jarzynski and Crooks equations) for alchemical free energy transformations and end-state (e.g., MM->QM or MM->ML) book-ending simulations
- advanced methods for network-wide alchemical free energy analysis of protein-ligand binding thermodynamic graphs with cycle closure and experimental constraints.
- Improved REMD structure reservoir options, including reservoirs for H-REMD and flexible reservoir temperature with T-REMD
- Gaussian accelerated MD (LIG-GaMD).
Improved Implicit Solvent Methods
- New tensor-based Poisson-Boltzmann solver backend in PBSA, with GPU acceleration, preconditioning, and matrix-free operation.
- Bundled LibTorch now ships in two versions (2.5.1 and 2.10.0), expanding CUDA support from 11.8 through 13.0.
- Implicit solvent/explicit ion solvent model (GBION). The parameters have been refined to handle DNA and protein-DNA complexes, most importantly the nucleosome.
- Added highly-accurate dSASA method for calculating the solvent accessible surface area in nonpolar solvation free energy calculations, as well as forces required for MD simulations.
Amber入門
1. Amberプログラム解説
Amberは生体分子系のシミュレーションを行うための複数のプログラムからなるソフトウェアパッケージです。用途で大別して入力準備プログラム、シミュレーションプログラム、結果解析プログラムから構成されています。それぞれの用途でも複数のプログラムが用意されています。それぞれの概要は以下の通りです。
入力準備プログラム
LEaP :
Amberのための新しい系を作ったり、既存の系を修正したりするための基本プログラムです。
LEaPは以前のバージョンのprep, link, edit, parmの機能を統合したものです。
ANTECHAMBER :
Antechamberファミリーソフトウェアパッケージのメインプログラムです。分子フォーマット変換、原子タイプ指定、電荷生成を自動的に行うため、標準的な核酸やタンパク質を多く含んだ系ならば、LEaPのインプットファイルを準備するのに役立ちます。
シミュレーションプログラム
ANDER :
エネルギー最小化と分子動力学法を行うための基本となるプログラムです。
エネルギー最小化ではエネルギー勾配の平均値が十分低くなるまで、エネルギーが下がる向きに繰り返し原子を動かすことによって構造を緩和します。
分子動力学法ではNewton運動方程式を積分することによって系の配置を生成します。
分子動力学法はエネルギー最小化より多くの配置空間をサンプルします。小さなエネルギー障壁を構造が超えることを可能にします。
配置は後々の解析のために、シミュレーション中に一定間隔で保存されます。熱力学積分を行って基本的な自由エネルギー計算をします。
より精巧な配置空間探索とモデリングの分子動力学法研究もSANDERモジュールを使って実行することができます。
これはいろいろな束縛条件を基本的な力場に加えることを可能にします。特にNMR構造詳細化に関与するタイプの計算のために設計されました。
PMEMD :
並列化効率と速度に関して最適化を図ったバージョンのsanderです。
プログラムの名称は”Particle Mesh Ewald Molecular Dynamics”に基づいており、この種のシミュレーションに限定されています。
入出力ファイルはsanderのものからほんの少しだけしか変更がありません。
NMODE:
準Newton Raphsonによる二次微分を利用したエネルギー最小化と振動解析のプログラムです。
NMODEは、系の多数の熱化学特性とノーマルモードを計算することができます。
他の機能として「 Langevin モード」(連続溶媒への粘性結合を含めたノーマルモード)を計算することが可能であり、エネルギー最小構造と同様に遷移状態を見つけるテクニックを備えます。
結果解析プログラム
PTRAJ :
MDシミュレーションによって(あるいは種々の他のソースから)生成された軌跡(trajectory)や座標を解析したり処理したりする汎用のユーティリティプログラムです。
・重ね合わせ
・座標の抽出
・結合長、結合角、二面角の計算
・原子の座標変位
・相関関数
・水素結合解析
等をの実行します。
同じ実行ファイルが名称をrdparmとして実行されるとき、prmtopファイルを検査したり修正したりすることが出来ます。
MM-PBSA :
連続溶媒モデルを利用した分子動力学シミュレーションからのスナップショットのエネルギー解析を自動化するスクリプトです。
2. Amber利用法概略
新たにAmberを利用されようとするお客様にAmberプログラム利用の流れを説明します(概略の説明ですので個々のプログラムの利用に当たっては個別プログラムのドキュメントを参照してください)。
どういった手順からAmberのシミュレーションを始めるべきかを理解することが第一の問題です。
まず最初にどのシミュレーションプログラム(sander, pmemd, nmode)を利用するかを理解する必要があります。
利用するプログラムが定まりましたら、次に全てのシミュレーションプログラムで必要とする情報を用意します。
全てのシミュレーションプログラムで必要とする情報は以下の通りです。
(1) 系の各原子の三次元座標
X線結晶解析、NMR分光法、モデルビルデングによってProtein Databank (PDB)で作成します。
これらのモデリングタスクの多くに対して、プログラムLEaPは実行プラットホームを供給します。
場合によっては他のプログラムを考慮しなければならないこともあります。
(2) "トポロジー": 結合, 原子名, 原子タイプ, 残基名, 電荷
これらの情報はamber8/dat/leap/prepディレクトリにあるデータベースから得ることが出来ます。
Amber8のマニュアルのChapter 2に詳細な記述があります。
そこには標準アミノ酸、N末端やC末端の荷電アミノ酸、DNA、RNA、通常の糖、等のトポロジーを含んでいます。
データベースはこれらの単量体ユニットのデフォルト内部座標を含んでいます、しかし通常、座標情報はPDBファイルから得られます。
(標準データベースで見いだされない)他の分子のためのトポロジー情報は、通常antechamberを使って利用者が作成し、利用者作成の「残基ファイル」に保存します。
(3) 力場: 系における結合、結合角、二面角、原子タイプなどのパラメータ
いくつかの標準的な力場パラメータはamber8/dat/leap/parmディレクトリに納められています。やはりAmber8のマニュアルのChapter 2に詳細な記述があります。
これらのファイルはタンパク質や核酸についてそのまま利用することもありますし、利用者が標準の力場に対して修正を行ったものを用意して利用することもあります。
(4) コマンド:シミュレーションのオプションと状態変数の指定
これらはmdinと名前を付けたsander, pmemd, nmodeの各プログラムの入力ファイルで指定します。
以上の情報は通常、LEaP、antechamberの入力ファイル作成プログラムを利用して準備します。
準備できましたらシミュレーションプログラム(sander, pmemd, nmode)を実行します。
実行が完了した結果、途中の経過等を解析プログラム(PTRAJ, MM-PBSA)で解析します。
これまでの流れは次の図のようにまとめられます。
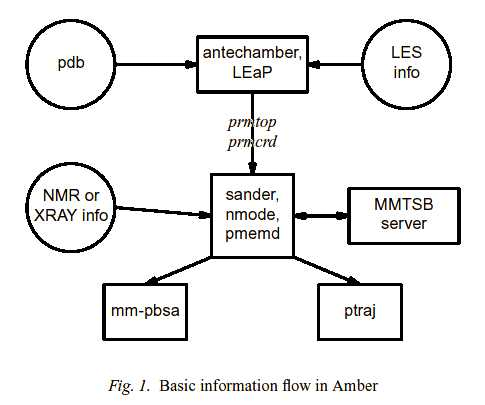
Amber8マニュアルより引用
Amber応用例
- Construction, MD Simulation, and Hydrodynamic Validation of an All-Atom Model of a Monoclonal IgG Antibody
J. Paul Brandt, Thomas W. Patapoff, and Sergio R. Aragon Biophys J. 2010 August 4 99(3):905-913.
HPCシステムズがビルドしたAmberの特徴
HPCシステムズは、Amberを研究の基礎として安心してご活用いただけるように、豊富な機能を盛り込んでコンパイルし、Amber規定の動作テストと速度検証を行って問題ないことを確認した上で、その報告書も同梱して納品しております。
詳細は以下のリンクをご覧ください。



お問い合わせ
平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)