
計算化学
ソフトウェアの選び方
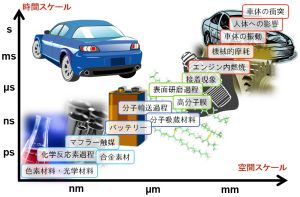
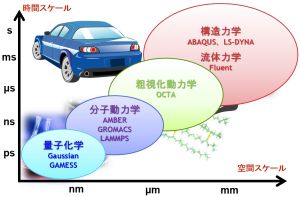
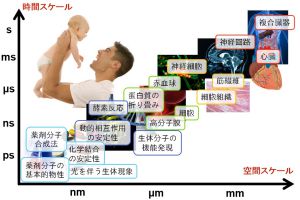
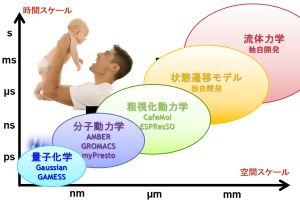
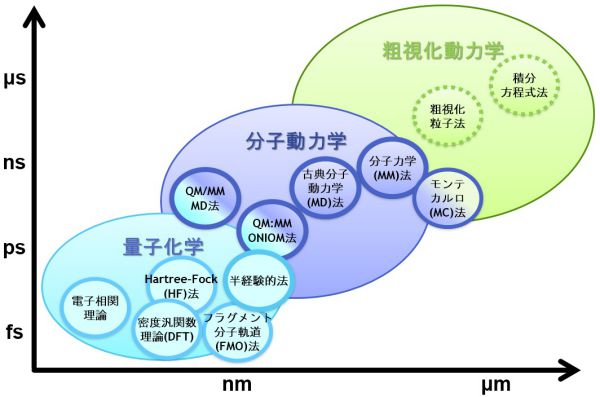
材料や現象のスケールと計算手法の関係
材料や現象のスケールと計算手法には関係があります。例えば、光デバイスは光と物質との相互作用を利用したものであり、物質の電子状態がその性質を決めています。したがって、電子状態が扱える量子化学シミュレーションや固体物理シミュレーションが利用されます。薬の性能については、薬が患部に到達するまでの過程、タンパク質と結合する過程、結合している状態などさまざまな現象が絡んでおり、それぞれに対して計算手法が異なってきます。図のように時間スケール・空間スケールに材料や現象を分類すると、シミュレーションに必要な計算手法がわかってきます。
量子化学シミュレーション
孤立した分子の構造や電子状態を扱うシミュレーション技法です。扱うことのできる状態は絶対零度・静止状態に制限されますが、電子状態が本質となる現象では個々の分子の特性がその多くを特徴づけているため、量子化学計算で十分対応できることが多いです。
Gaussianは量子化学ソフトウェアのデファクトスタンダード
量子化学シミュレーションにはGaussianがおすすめです。世界的に広く利用されているソフトウェアで、量子化学研究者が要求する多くの機能が利用できます。計算速度が速く、また専用の可視化ソフトウェアGaussViewもあるため、非常に操作がしやすいのが特徴です。
特色ある機能を搭載したソフトウェア
その他にもさまざまなソフトウェアがあり、特色ある機能が搭載されています。
- GAMESS – プログラム構造が比較的簡単で、量子化学の基礎理論開発によく利用されている。
- Molpro – 高精度電子相関計算や励起状態計算に特化した機能が特徴的。
- NWChem – RI-MP2に対応 / 反応経路探索法としてNEB(Nudged Elastic Band)法が利用可能。
分子動力学シミュレーション
古典分子動力学では電子状態を扱わない代わりに、原子の動きや温度の影響を取り入れたシミュレーションが可能です。
扱う材料や知りたい物性に合わせてソフトウェアを選択する必要があります。
生体分子
AMBERや GROMACSでは生体分子向けにチューニングされた力場(AMBER力場など)が利用できます。生体分子シミュレーションでよくおこなわれる解析手法もこれらのソフトウェアに搭載されています。計算速度を重視するのであれば、世界最速を目指しているGROMACSがおすすめです。
電解質などの溶液系
分子の種類によっては適切な力場が用意されていないこともあります。力場パラメータを文献調査し、その力場が扱えるソフトウェアをAMBER, GROMACS, LAMMPSなどから選択して使用します。必ずしも文献が見つかるとは限らず、分析のための指針を得ることを目的として必要に精度は求めず、傾向を調査するためと割り切って、AMBER力場を使用する研究者もいます。
樹脂などの高分子材料
材料系を作成して静的状態を観察するだけであればAMBERやGROMACSでも可能ですが、引っ張る・曲げるなどの試験には力学的操作が可能なLAMMPSがおすすめです。
金属材料やカーボン材料
LAMMPSでは金属材料やカーボン材料向けの力場が利用できます。引っ張る・曲げるなどの力学的操作が可能です。
化学反応を伴う現象
化学反応は電子が絡む現象であるため、量子化学と分子動力学を組み合わせた量子分子動力学計算に対応したソフトウェアを選択することになります。AMBERでは着目する部分だけを量子化学で扱うQM/MM法が利用できます。またCPMDでは全系を量子化学で扱うCar-Parrinello法が利用できます。
古典分子動力学ではns~μsのスケールの計算が可能ですが、量子分子動力学は非常に計算コストが高く、~100ps、条件によっては~数psの計算に制限されるため、シミュレーションを始める前に現象の本質を見極め、適切にモデル化する必要があります。
お問い合わせ
平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)