
Software
計算化学
化学系計算手法の特徴比較
ある物質・物体や現象を計算機でシミュレートしたいと考えた場合、まずその研究対象がどのくらいの時間スケール、空間スケールに位置しているのかを把握せねばなりません。これは、研究対象のスケールによって、適切なシミュレーション理論・ソフトウェアが異なってくるためです。
下図はそれを大雑把に示したもので、例えばタンパク質と薬分子のドッキングの初期過程を計算するためには、nm、nsオーダーの現象の記述に適した理論とソフトが必要であることを示しています。
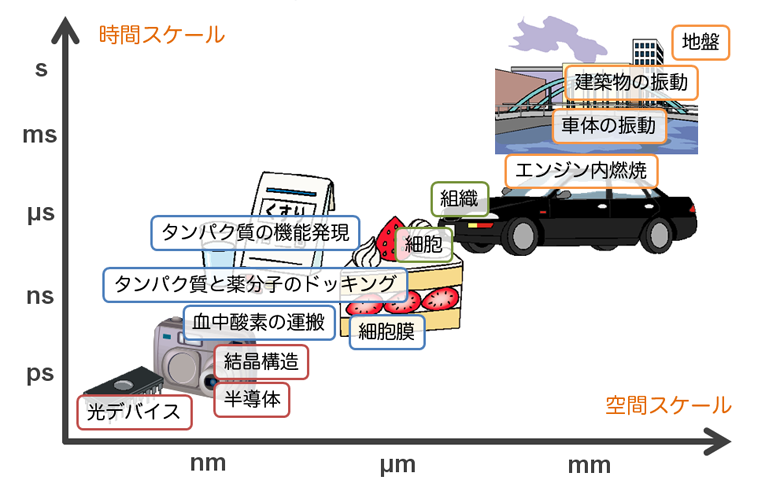
対象となる系のスケールに応じた計算手法は、大まかに以下のように分類できます。建築物強度や風洞解析のようなマクロな現象であれば、ABAQUSやFluentのようなソフトが適切となりますし、逆に、化学反応や半導体の電気伝導のようなミクロな現象であれば、Gaussianのような量子化学計算ソフトやVASPのような固体物理計算ソフトが役立つでしょう。近年は、計算機性能の向上に後押しされるように、ミクロな現象のシミュレーションからマクロな現象の本質を読み解く研究が盛んに行われており、特に光が関与する現象は有機ELや太陽電池の製品開発などの工業的見地からも計算機シミュレーションが活用される機会が増えてきています。
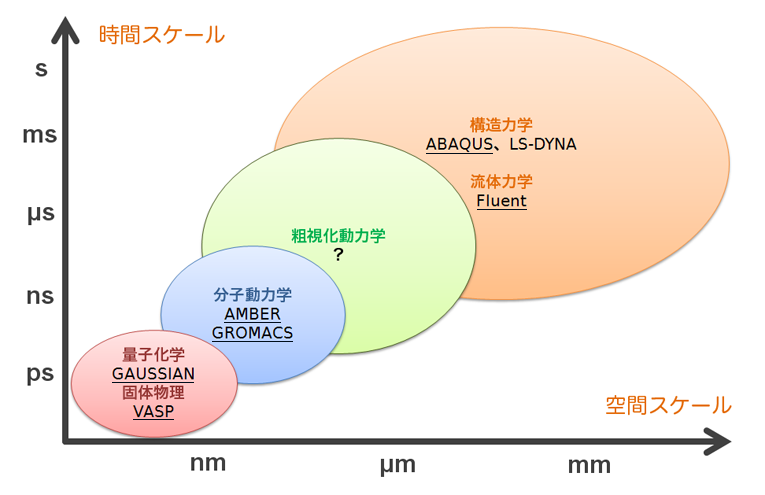
ミクロな現象の計算手法としては、大きくわけて、量子化学・固体物理・分子動力学の3つが挙げられます。それらの特徴を下の表にまとめました。
これらの手法は、究極的に言えば、ある共通の方程式を出発点にしています。ただ、その方程式を現実的な時間で解くことは事実上不可能であり、その不可能を可能にするためにどのような近似を導入したかが各手法によって異なっています。その近似の精度は、量子化学 > 固体物理 > 分子動力学であり、それに伴い計算コストも、量子化学 > 固体物理 > 分子動力学となります。
実際には、対象となる系のスケールとこれらの長所・短所などを天秤にかけながら研究を進めていくことになりますが、可能であれば、結果として見られる長所・短所だけで判断するのではなく、その手法の原理から理解した上で使い分けていくことが望ましいです。
| 量子化学計算 | 固体物理計算 | 分子動力学計算 | |
|---|---|---|---|
| 主なソフト | Gaussian、GAMESS | VASP、WIEN2k | AMBER、Gromacs |
| 最大の「売り」 | 高精度・高信頼性 | 大規模固体系が扱える | 分子を動かせる |
| 主な近似事項 | 断熱近似、軌道近似 | 左記+V核-電子の近似 | 電子は顕に考慮しない |
| 計算コスト | 高価 | 比較的安価 | 安価 |
| 得意な系 | 小規模孤立系 (気相分子) | 周期系 (理想的結晶構造) | 大規模系 (生体分子等) |
| その他の利点 | 化学反応が扱える 水素結合、vdW力の記述 様々な物性値が計算可能 | 様々な物性値が計算可能 ダイナミクスも一応可能 | 溶媒効果の評価 温度・濃度の考慮 |
| 弱点 | 比較的小規模系のみ ダイナミクスは実質上困難 | 非周期系には弱い 化学反応は難しい | 結果が力場に大きく依存 化学反応は扱えない 物性値計算は手動 |
| 現在主流の計算手法 | ハイブリッドDFT | (メタ)GGA-DFT | MM(分子力学) |
| それを超える精度の計算 | MP2法、CCSD法、SAC-CI法など多数 | vdW補正DFT法など | QM/MM法など |
実際には、対象となる系のスケールとこれらの長所・短所などを天秤にかけながら研究を進めていくことになりますが、可能であれば、結果として見られる長所・短所だけで判断するのではなく、その手法の原理から理解した上で使い分けていくことが望ましいです。
お問い合わせ
平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)