
Software
計算化学
新しい材料を化学反応でつくる
1. はじめに
古代から現代まで我々人類はさまざまな材料物質を化学反応から作り出し、文明を発展させてきた。かつては経験と偶然の中から反応の仕組みを理解し、試行錯誤して産業に役立ててきた。しかし、膨大な可能性の中から経験と偶然だけで正解を見つけることは難しく、加えて今日では製造コストや環境負荷など、さまざまなことに注意を向けなければならず、戦略的に材料を生み出す技術が求められている。
反応設計で考えなければならないこと
- 反応速度や収率
- 材料のコスト
- リサイクル可能な副産物
- 安全性
- 装置のメンテナンス性 などなど
2. 化学反応は反応経路で理解できる
化学反応の仕組みを考えるために、アンモニア(NH3)が水(H2O)と反応して、アンモニウムイオン(NH4+)ができるという、単純な反応を考えてみよう。
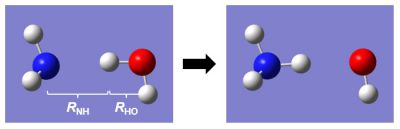
※この分子系は説明のしやすさのために用いているだけであり、
実際のエネルギーダイアグラムとは異なるため注意。
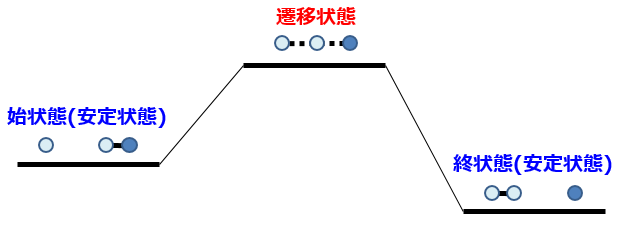
N-H間距離とH-O間距離に着目して反応の様子を見てみると下図のように示される。すると、化学反応とは高さ(=エネルギー)の低い谷の部分を通って分子が変形すること、と理解することができる。
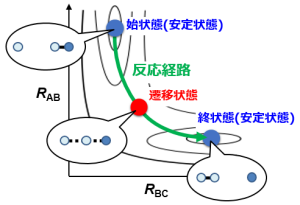
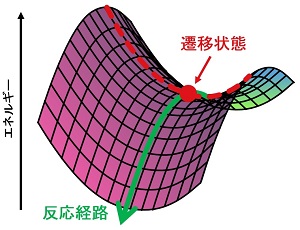
分子が小さく反応が単純であれば、このように図示して説明することは簡単であるが、実際に多くの研究者が扱いたい反応系は100原子以上。数学的には何百次元ものエネルギーマップのなかから1本の反応経路を見つけることに相当し、非常に難易度の高い問題なのである。
3. 量子化学計算で反応経路を予測する
研究者がまず知りたいのは、反応の始状態、遷移状態、終状態の3つに対する構造とエネルギーである。Gaussianをはじめとする多くの量子化学計算プログラムには、分子構造を最適化する機能が実装されているのでこれを使えばよい。始状態や終状態は安定状態であるため、比較的簡単に構造を見つけることができる。
やっかいなのは遷移状態の構造である。この構造は反応経路の方向に沿った変形に対しては不安定なため、ユーザが遷移状態の構造に極めて近い構造をインプットさせない限り、うまく最適化構造は見つけられない。答えがわからないから計算で予測してもらいたいのに、これでは非常に困る。
4. 考えうるすべての反応経路を全探索する
GRRM (Global Reaction Route Mapping) 法という方法がある。これは秀逸な方法で、与えた構造の周りを測量しながら、遷移状態がある方向へ分子を変形させ、これを繰り返すことによって、考えうるすべての反応経路を全自動で探索する方法である。
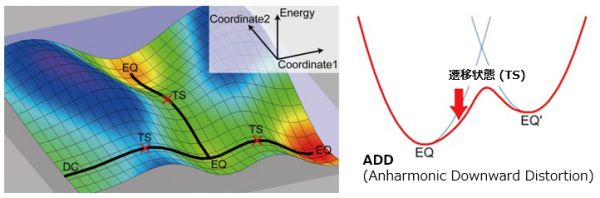
調和関数で予測されるエネルギー面よりも下方のエネルギー値をとる経路上に遷移状態はあるとする
ADD(Anharmonic Downward Distortion)に基づいて、反応経路の全探索が行われる。
ユーザは知りたい反応系の分子を1つインプットするだけでよい。後は待っているだけですべての反応経路が見つかる。すべての反応経路のため、ユーザの知りたい反応経路はかならずその中に見つかる。正しい答えが確実に求まるのである。ユーザの試行錯誤は一切いらず、計算中に別の仕事ができることも大きなメリットである。
しかしデメリットもある。この方法では数学的に考えうる経路をすべて求めようとし、研究者から見て化学的にありえない経路まで探索してしまうために、非常に計算時間がかかってしまうのである。探索範囲を限定するなどさまざまな改良がおこなわれており、今後が楽しみである。
GRRM法が利用可能なソフトウェア:
- GRRM
5. 化学センスを活かして、半自動で反応経路を予測する
研究者の経験とそれに基づく勘には素晴らしいものがあり、反応経路がある程度の想像できてしまうことがある。そのうえで計算機に問いかけたいのは、そのような反応機構で反応するとしたとき、正確にはどんな構造になりどんなエネルギーになるのか、そして考えた反応経路のうちどれが優位な反応経路なのか。そのような場合には、NEB (Nudged Elastic Band) 法 や String 法が使えるかもしれない。
NEB法やString法ではユーザは適当に始状態や終状態、必要に応じて中間状態の構造をインプットするだけでよい。計算の仕組みは、エネルギーマップ上にたらしたひも(仮の反応経路)が、真の反応経路に向かって最適化されていく、そんなイメージである。ひも上の各分子構造の計算が並列に実行できるため、計算機のパワーを余すことなく使うことができ効率的である、というメリットもあり、この高効率さゆえ、分子のサイズが100原子以上になっても比較的簡単に反応経路を見つけることができる。
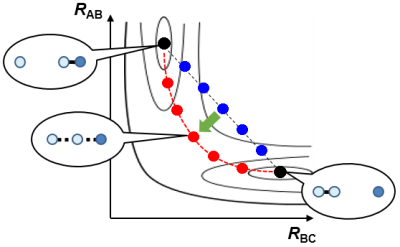
まずユーザの入力したインプット(この図では始状態と終状態の2点のみ)の間を
仮の反応経路(青)で結ぶ。仮の反応経路上の各分子構造(丸印)の位置を、
ひもで繋がれた隣り合う構造の動きを考慮しながら最適化していくと、
仮の反応経路が正しい反応経路(赤)に向かって最適化されていく。
NEB法やString法が利用可能なソフトウェア:
- NWChem
- Reaction plus
6. まとめ
反応経路の探索の難所は、遷移状態構造を見つけることにある。この遷移状態構造をユーザが手探りで見つけるのは神業であるが、GRRM法を使えばそのような苦労は一切なく、待っているだけで正しい反応経路が見つかる。
一方、ある程度反応機構に想定が付いている場合には、NEB法やString法を使うことで、比較的短時間で反応経路を見つけることができる。場面に応じて手法を使い分けるのがよさそうだ。
お問い合わせ
平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)