
Science as a Service
Software
Software
計算化学 ソフトウェア
Gaussian 日本語マニュアル
モデル化学
方法と基底関数系の組み合わせにより,Gaussianでのモデル化学を指定,つまり理論レベルを指定します。全てのGaussainジョブで方法と基底関数系を両方とも指定しなければなりません。これには通常,インプットファイルのルートセクションに個別にキーワードを2つ指定します。ただし,方法キーワードによっては基底関数系の選択もそれ自身に含まれているものもあります。
次の表にGaussianで利用可能な方法について,その方法を用いることができるジョブタイプごとに示してあります。ただし,この表での最適化,振動数,分極率計算は解析的な計算のみであり,数値的な計算についてはチェックされていない方法でも利用可能なものがあることに注意してください(詳細については各キーワードの説明を参照してください)。
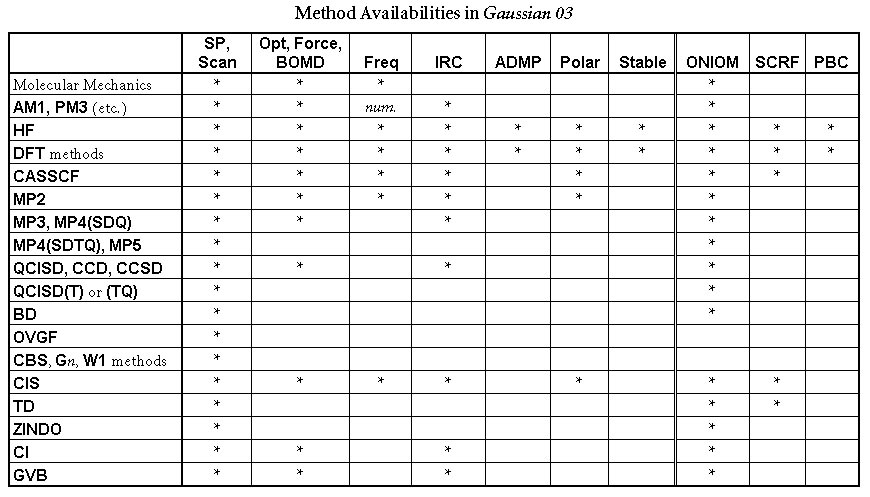
方法キーワードが指定されなかった場合には, HFとされます。ほとんどの方法では,各方法論の接頭にRをつけると閉殻制限波動関数に,Uをつけると非制限開殻波動関数に,ROをつけると制限開殻波動関数となります。たとえば,ROHF, UMP2, RQCISDのようにします。ROはHartree-Fock, 全密度汎関数法,AM1, MINDO3, MNDO, PM3半経験的エネルギー・グラジェント(勾配),MP2エネルギー計算でのみ可能です。ただし解析的ROMP2グラジェントは今のところ利用できません。
通常,指定できる方法キーワードは一つだけであり,方法キーワードを複数指定すると奇異な(bizarre)結果となるでしょう。しかし,この例外として:



CONTACT
お問い合わせ
お客様に最適な製品をご提案いたします。まずは気軽にお問い合わせ下さい。
075-353-0120
平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)