
Software
計算化学 ソフトウェア
Gaussian入門メールニュース:
Gaussian計算エラー対処・虎の巻
「計算がエラーで止まってしまった!でもどうすればいいかわからない。」
「結果が何かおかしい。どこがいけなかったのだろうか?」
といった疑問の解決に少しでもお力添えするべく、よくあるエラーを中心に、直接的な対処法から簡単な理論的背景まで含めて解説いたします。
第11回配信: エネルギーSCF手続きに関するエラー〔1〕
HF計算やDFT計算におけるエネルギーの未収束による計算停止は、恐らく、Gaussian習得レベルを問わず、ユーザにとって最も頭を悩ませるエラーの1つだと思います。なぜなら、最終的にユーザが目にする情報は「収束に失敗しました」という1種類のエラーメッセージですが、収束失敗に至った根本の原因は多種多様で、その対処法も多岐にわたるからです。そこで、このエラーについては今回と次回の2回に分けて解説します。
(11-1) まずは人為的ミスを疑おう
HF/DFT計算におけるエネルギーSCF手続きが収束に至らなかった場合、以下のようなエラーメッセージとともに、Overlay 5で計算が停止します。
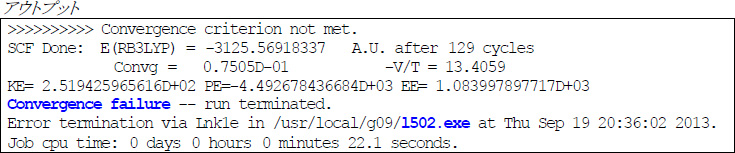
教科書によっては、こういう場合の解決策として、SCF=(MaxCycle=N) キーワードに大きい N を指定して、SCF繰り返し手続き回数上限を増やすことが挙げられているかもしれません。しかし、Gaussian 09のデフォルト設定ではN=128であり、これは通常の分子では十分大きい数字です。筆者の経験上、128回も繰り返し計算してもエネルギー(または分子軌道)が収束しない場合、単純に N を増やしてみても事態は好転しないことがほとんどです。
実は、エネルギー(分子軌道)が収束しない場合も、大抵は単純な人為的ミスによるものであり、まず行うべきことは計算入力条件の確認です。不適切な分子構造指定や不適切な基底関数指定をインプットで与えると、大抵は直ちに計算が異常停止するのですが、時には不適切ながらも計算自体は続行されることがあります。エネルギーが収束しない場合の多くは、このケースにあたります。まずは、第3回配信や第7~10回配信の内容を参考にしながら、インプットに不適切な箇所がないか、よく確認しましょう。
とはいえ、SCF収束が難しいという分子系は確かに存在し、そのような計算では人為的なミスがなくてもエネルギーが収束しないこともよく起こります。以下ではその場合の対処法を紹介していきます。
(11-2) SCF収束状況の確認
具体的な対処法に入る前に、HF/DFTエネルギー(および分子軌道)を求めるためのSCF手続きについて少し復習しておきましょう。HF/DFTエネルギーを求めるには、以下のRoothaan方程式を解く必要があります。
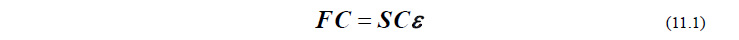
ここで、行列 F と S がこの方程式のインプットパラメータで、方程式の解として C と ε が求められます。さて、本来ならば、行列 F と S が与えられた時点でこの方程式は直ちに解けるはずなのですが、現実はそうではありません。というのも、F が(本来答えとなる)C に依存する行列であるため、C がわからないと F を決めることができないからです。そこでまず、C の初期値 C0 を適当に与えて F を計算します。この初期値 C0 のことを「初期軌道」(initial guess)と呼びます。その F を用いて方程式を解くと C と ε が求まるのですが、このままでは、求まった C と、Fの中に含まれている C は一致していません。そこで、新たに求まった C を用いて F を更新し、再び方程式を解いて、そこから得られる C を用いて F を更新し……という手続きを C がもはや変わらなくなるまで繰り返して、最終的な解を得ます。この手続きは「自己無矛盾な場」または「自家無撞着な場」(self-consistent field; SCF)の方法と呼ばれます。
したがって、順調なSCF手続きの中では、現在の C と更新後の C の「差」に相当する値が少しずつ0に近づいていき、それが一定の閾値(Gaussianでは通常10−8)より下回れば収束と見做されます。この「差」はアウトプット中では「RMSDP」という値で表されますので、順調に収束に向かっているかどうかは、この値をチェックしていけばわかります。下の例では、12回目のSCF手続きでRMSDPが1.41×10−9となり、閾値10−8を下回ったため、収束に至っています。

ただし、RMSDPの値は、インプットで#P(詳細出力)を指定しないと出力されませんので、ご注意ください。
(11-3) エネルギーが収束しない場合の対処法
人為的なミスがないにも拘らずエネルギーが収束しない場合、残念ながら、決定的な対処法があるというわけではありません。以下に、幾つかの対処法を列挙しますが、実際には、これらの対処法をひとつずつ、あるいは複数の方法を組み合わせて、試していくしかありません。
◆Restricted Open-shell (RO)-SCF法の代わりに、Unrestricted (U)-SCF法を採用する
開殻分子系をROHF法/RODFT法で計算しようと考えている場合、UHF法/UDFT法で代替できないか検討してみましょう。一般に、ROHF/RODFT法よりUHF/UDFT法を用いた方が良い収束性をもたらします。(なお、Gaussianのデフォルト設定はUHF/UDFT法です。)
◆N 電子開殻分子の場合、(N+1)電子閉殻分子の計算結果を初期軌道に採用する
奇数電子系(特にアニオンラジカル)の場合に有効な対処法ですが、目的の系(N 電子系)より1個電子を増やした (N+1)電子系のエネルギー計算を行い、そこで得られた分子軌道を目的の計算の初期軌道に採用すると、上手く収束する場合があります。
具体的手順としては、(N+1)電子系の計算結果をchkファイル(ここではファイル名をaaa.chkとする)に保存した後、N 電子系計算のインプットファイルで「%Chk=aaa.chk」および「Guess=Read」を指定します。(また、第7回配信も併せてご参照下さい)
◆低精度基底関数の計算結果を初期軌道に採用する
一般に基底関数の質を良くしていくと、収束も難しくなる傾向にあります。そこでこの性質を逆手に取り、一旦、基底関数の質を落としてエネルギー計算を行い、そこで得られた分子軌道を目的の計算の初期軌道に採用すると、上手く収束する場合があります。
具体的手順は上記とほぼ同じで、本計算のインプットでは%ChkとGuess=Readを指定します。
◆SCF=(MaxCyc=N) の N を増やす(繰り返し回数上限を増やす)
冒頭で、この N を増やしても事態はほとんど好転しないと書きましたが、まれにこの対処で収束することもあります。特にRMSDPの値が僅かずつながらも収束に近づいている場合は有効かもしれません。
◆SCF=(Conver=N) の N を減らす(収束閾値を甘くする)
RMSDPの値が10−8や10−7の辺りを行ったり来たりしている場合は、Conver=7やConver=6を指定して、収束閾値を10−7や10−6に設定してやれば、それらの値でも直ちに収束と見做されます。通常、Converの値は多少甘くしても、実用上さほど問題ありません。
ただし、Conver=4以下にはすべきではありません。また、励起状態の計算など、ある程度のSCF解の収束性が求められる場合は、Conver=5以下にはすべきではありません。
◆SCF=Fermi、SCF=QC を指定する
SCF手続きのアルゴリズムはひとつではありません。Gaussianのデフォルト設定ではDIISアルゴリズム(SCF=DIIS)が採用されていますが、このアルゴリズムで収束しない場合は、別のアルゴリズムを用いてSCF手続きを行うと、スムーズに収束する場合があります。
Fermi法は、DIIS法で収束しなかった場合に、まず試すべきアルゴリズムです。また、時間はかかるものの、収束点到達への信頼性の高いQC法を採用するのも有効であり、DIIS法やFermi法で収束しなかった場合は、試してみる価値があります。(ただし、ROHF/RODFT法では、SCF=QCは指定できません。)
◆SCF=XQC、SCF=CDIIS、SCF=SD を指定する
Fermi法やQC法を用いても収束しなかった場合、これらのアルゴリズムを用いることで収束する場合があります。ただし、あまり期待してはいけません。
◆SCF=(VShift=N) を指定する
非占有軌道を人為的に高エネルギー側へシフトさせ、占有軌道と非占有軌道のエネルギー差を大きくしてやると、SCF手続きが収束する場合があります。N はエネルギーシフト量(ミリハートリー単位)を表し、通常、N=100~1000程度の値を指定します。(デフォルトでは N=100です)
単独で用いてもよいですし、他の収束アルゴリズム(SCF=FermiまたはSCF=QC)と組み合わせて用いることもできます。
◆占有軌道と非占有軌道を適切に入れ替えて、初期軌道に採用する
得られた分子軌道を解析し、ある占有軌道とある非占有軌道を適切に入れ替えたものを初期軌道として再計算を行うと、上手く収束する場合があります。
これについては、次回の第12回配信で詳しく解説します。
◆そもそもHF/DFT法では計算不可能
解離過程のポテンシャルカーブをHF/DFT計算で描いていこうとすると、解離が進むにつれて、エネルギーの収束性が悪くなったり、あるいは収束しても信頼できない結果となったりすることがあります。これは、(閉殻分子同士への解離などの特殊なケースを除き)平衡分子構造から離れた構造では、HF法やDFT法が、そもそも原理的に計算不可能となるためです。
そのような場合、CASSCF法に代表される「多参照電子相関理論」と呼ばれる手法を用いた計算を検討する必要があります。ただしその詳細については、本メールニュースの難易度の範囲を大きく超えてしまいますので、お知り合いの量子化学研究者の方にお尋ねするか、弊社の有償サポートサービスをご利用ください。
今回の内容は以上です。次回(第12回配信)では、「エネルギーSCF手続きに関するエラー〔2〕」を解説いたします。
メールニュース会員募集中!
Gaussian入門者の皆様に、Gaussianを使いこなして高度な計算化学者に飛躍していただけるよう、Gaussianに関する様々な情報を発信しております。メールニュースの配信をご希望の方は、以下のフォームよりお願いいたします(お問い合わせ内容の項に「Gaussian入門メールニュース配信希望」とご記入ください)。
メールにてお申し込みされる方は、以下の必要事項をご記入の上、送信してください。
- ◆電子メール件名 : Gaussian入門メールニュース配信希望
- ◆記入項目:
1. 氏名 :
2. E-mail:
3. 法人名/部署名:
4. 住所:
5. 電話番号:



お問い合わせ
平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)