
Software
計算化学 ソフトウェア
Amber
Amberは米カリフォルニア大学のP. A. Kollmanらのグループにより開発された、生体分子の分子動力学(MD)計算のための力場群であり、またこれらの力場をシミュレーションするためのMDプログラム群です。現在は、米ラトガース大学のD. A. Caseらによるグループにより維持・管理されています。プログラム群は、入力準備プログラム・シミュレーションプログラム・結果解析プログラムに大別されます。GPUのみならず、最新Xeon Phiプロセッサーにも最適化されたAmber16が2016年4月にリリースされました。
Status
https://ambermd.org/AmberPatches.php
Amber18 Updates (April, 2018)
update.1: Adds an error message to pmemd when users run REMD and the box sizes are not the same across all replicas.
update.2: Fix in pmemd the neighbor pairs inside the pH and redox potential dimensions on multidimensional REMD simulations.
update.3: This update fixes the following:
Corrects REMD error trap for different box sizes in pmemd to account for non-periodic systems.
Bugfix in pmemd to properly restart MultiD-REMD simulations with two or more pH, redox potential or temperature dimensions.
update.4: Major update to pmemd.cuda, fixing various bugs. See the GPU Patches page for a full description.
update.5: Adds support for MC and Berendsen barostats in order to run the benchmark suite in the midpoint code. Additionally improves dynamic memory allocation.
update.6: Fix rare non-reproducibility issue in pmemd.cuda.
Version 18 (April, 2018)
update.1: Fix compilation of cpptraj with Intel compilers on older systems.
update.2: Adds an error message to sander when users run REMD and the box sizes are not the same across all replicas.
update.3: Fixes typo in genremdinputs.py’s help message, and corrects E,pH,T-REMD and pH,T-REMD tests affected by Amber18’s update.2.
update.4: Fixes test for boost libraries need by packmol_memgen.
update.5: Fixes a memory leak in NAB/libsff programs.
update.6: Updates the sample rism1d mdl files, to reflect up-to-date ion parameters.
update.7: Updates the configure script to allow cuda version 9.2 to be used.
update.8: This update fixes the following:
Corrects REMD error trap for different box sizes in sander to account for non-periodic systems.
Bugfix in sander to properly restart MultiD-REMD simulations with two or more pH, redox potential or temperature dimensions.
Corrects one Amber test affected by the bugfix.
Fixes typos in fitpkaeo.py and genremdinputs.py.
update.9: Companion patch to the Amber update.4, updating the configure script (which is a part of AmberTools). This patch only affects pmemd.cuda.
特長
溶媒理論3D-RISMとハミルトニアンレプリカ交換法が実装されたことで、計算の精密さが増し、かつより大規模な系での計算が可能となりました。また、構造立脚型の創薬に用いられています。細胞内の環境を再現し、結合自由エネルギーを定量的に求めることができます。GAUSSIANとの連携も可能です。
Amberの基本機能
- 自由エネルギー摂動法や熱力学的積分法などによる自由エネルギー計算
- 溶媒中での自由エネルギー計算
- エネルギー極小化計算
- MDシミュレーション
- 原子間距離や角度の算出
- 温度因子(B-factor)の算出
- 構造変化や構造の揺らぎをみる根平均2乗偏差(RMSd)の算出
関連情報
- Amber入門
- Amber応用例
- Amber検証日記 (2010年7月~2011年7月)
- Amber12 ユーザーマニュアル (PDF:231KB)
- QMMM plus 2 – Amberをもっと便利にするための便利ツール (弊社製品)
- Amber14の変更点
- Amber12の新機能
- Amber11の新機能
- Amber10で追加された機能
- 参考書籍:Essentials of computational chemistry: theories and models (著書:CJ Cramer) (外部リンク)
- 参考書籍:コンピュータ・シミュレーションの基礎 (著者:岡崎 進・吉井 範行) (外部リンク)
- 参考書籍:Amberによる生体高分子シミュレーション入門 (著者:Amber研究会) (外部リンク)
- 参考書籍:すぐできる分子シミュレーションビギナーズマニュアル (著者:長岡 正隆) (外部リンク)
- HPCシステムズの計算化学ソリューション – コンサルティング・受託計算・セミナー
ベンチマーク
ピックアップ情報:NVIDIA V100 による AMBER16 ベンチマーク結果
-
- PME-Cellulose_NPT on V100s PCIe
-

-
- PME-Cellulose_NPT on V100s SXM2
-
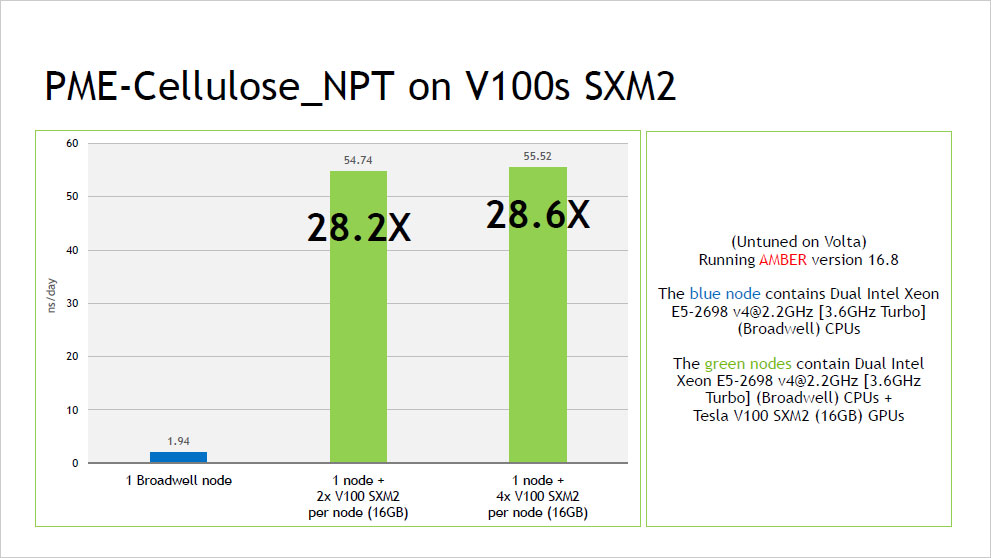
-
- PME-Cellulose_NVE on V100s PCIe
-
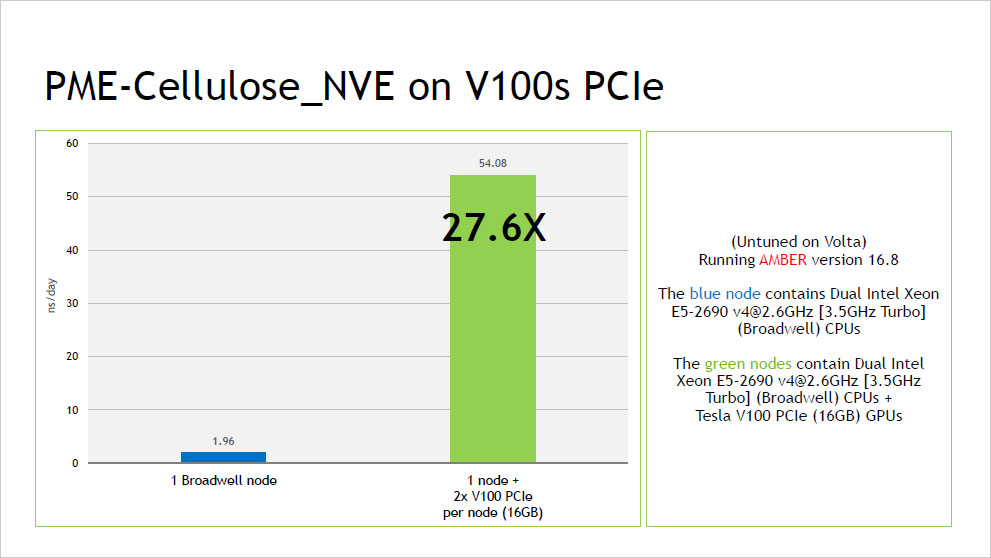
-
- PME-Cellulose_NVE on V100s SXM2
-
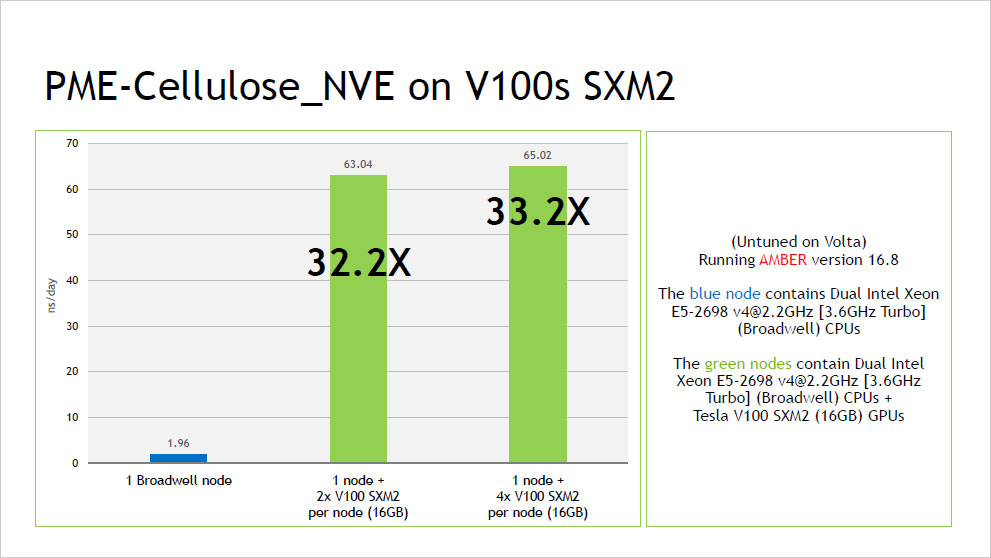
-
- PME-FactorIX_NPT on V100s PCIe
-
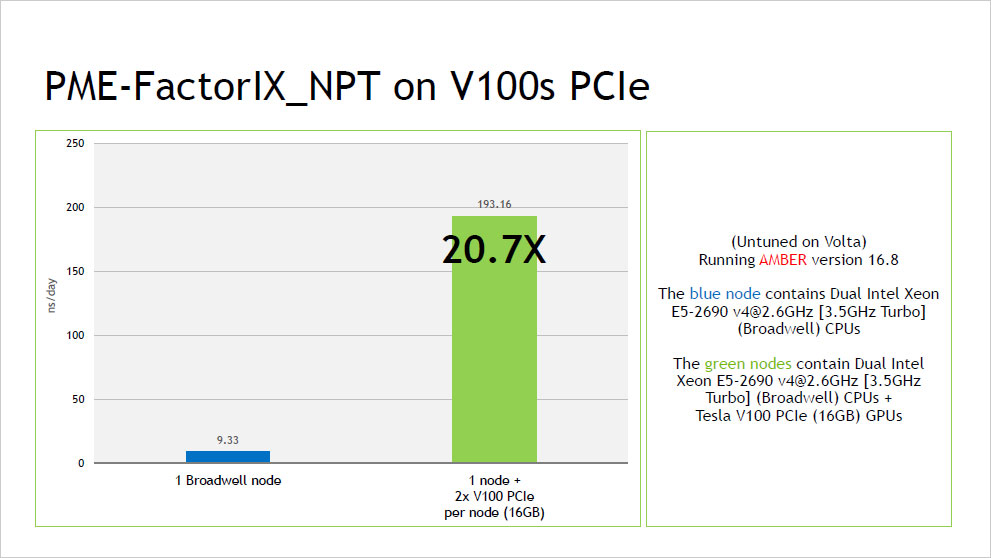
-
- PME-FactorIX_NPT on V100s SXM2
-
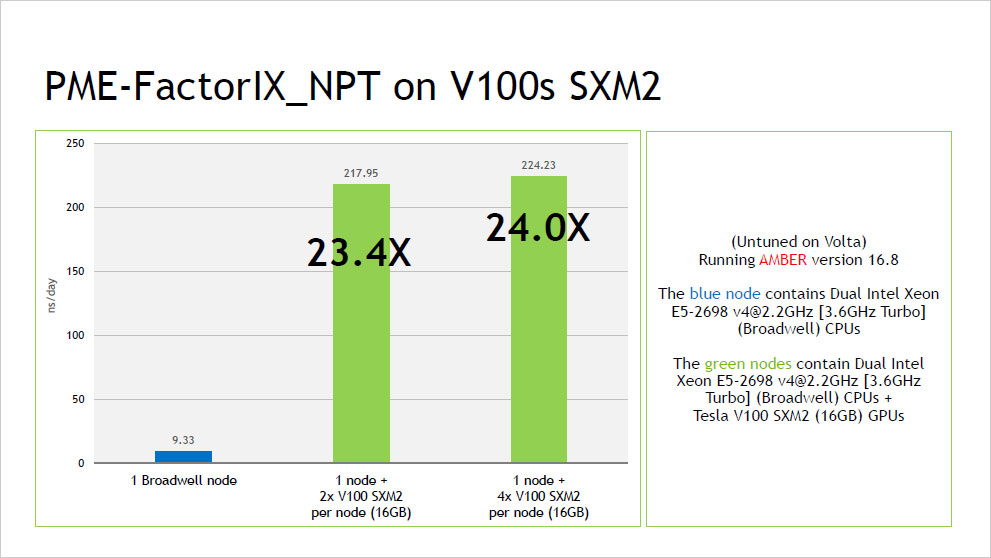
-
- PME-FactorIX_NVE on V100s PCIe
-

-
- PME-FactorIX_NVE on V100s SXM2
-
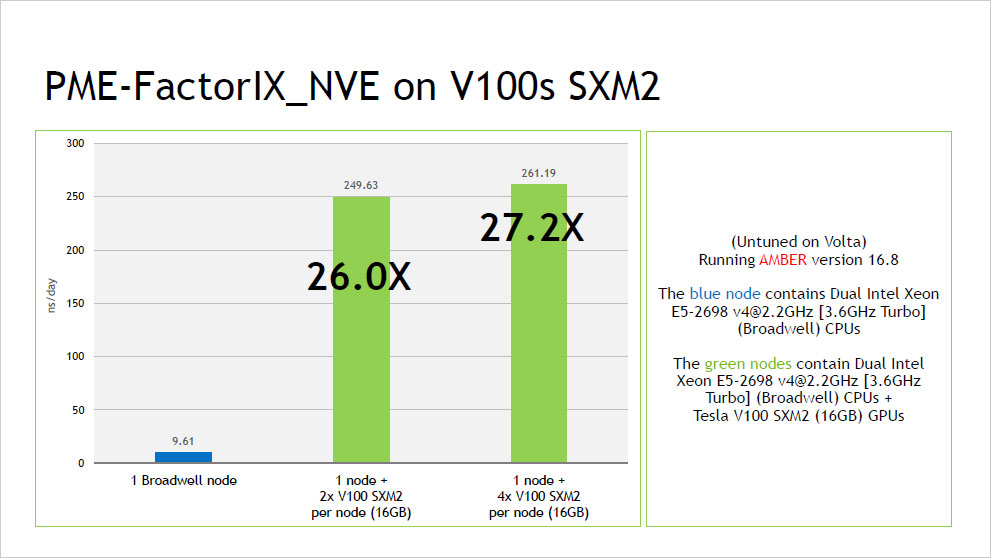
- NVIDIA V100 による AMBER16 ベンチマーク結果 (2017年10月:NVIDIA社提供資料) (PDF:352KB)
- NVIDIA P100 による AMBER16 ベンチマーク結果 (2017年2月:NVIDIA社提供資料) (PDF:360KB)
ベンチマーク情報
- NVIDIA K40・K80 Amber14 ベンチマーク比較 (2015年7月10日)
- NVIDIA K80 ベンチマーク結果 (2015年2月17日)
- インテル® Xeon® プロセッサー E7-8800 v2 ファミリー ベンチマーク結果 (2015年2月13日)
- インテル® Xeon® プロセッサー E5-2600 v3 ファミリー ベンチマーク結果 (2014年9月9日)
- インテル® Xeon® プロセッサー E5-2600 v2 ファミリー ベンチマーク結果 (2013年9月11日)
- NVIDIA K10でのAmber 12 GPUベンチマーク結果 (2013年1月7日)
- インテル® Xeon® E5-2600シリーズ 8ノードクラスターでのAmber 12ベンチマーク (2012年10月5日)
- インテル® Xeon® E5-2670 ベンチマーク結果 (2012年3月7日)
- CUDAでAmberを高速化! (2009年)
Amber14の変更点
・シミュレーションを準備・解析するための手続きの改善
Improved workflow for setting up and analyzing simulations
・cpptrajの機能拡張。Grid Inhomogenous Solvent Theory (GIST) の導入
Greatly expanded cpptraj functionality, including support for the Grid Inhomogenous Solvent Theory (GIST)
・新しい半経験的Born-Oppenheimer分子動力学法 (SEBOMD) 機能
New capability for semi-empirical Born-Oppenheimer molecular dynamics (SEBOMD)
・熱力学的積分法による絶対自由エネルギー差計算のための新しい手法
A new method to compute absolute free energy changes using a thermodynamic integration scheme (EMIL)
・多次元レプリカ交換分子動力学法 (sander, pmemd, pmemd.cuda)
Multi-dimensional Replica Exchange molecular dynamics in sander, pmemd, and pmemd.cuda.
・可能な限りピアツーピア通信を利用することで並列性能とスケーラビリティが大幅に向上 (pmemd.cuda)
Improved performance for pmemd.cuda and vastly improved parallel performance and scaling, using peer-to-peer communications when available.
・リファレンスマニュアルの再編成
Completely reorganized Reference Manual
・scaled分子動力学法の実装 (sander, pmemd)
Implementation of scaled Molecular Dynamics in sander and pmemd
・QM/MM計算において、アダプティブ法によるQM領域に対応
QM/MM calculations can have adaptive quantum regions
・Adaptive buffered force-mixing QM/MM法
Adaptive buffered force-mixing QM/MM calculations can be run
・implicit溶媒モデルや露わな溶媒におけるpH一定の分子動力学法 (sander, pmemd)
Constant pH molecular dynamics is now supported in implicit and explicit solvent in both sander and pmemd.
・pH一定レプリカ交換分子動力学法 (sander, pmemd)
Constant pH replica exchange molecular dynamics has been implemented in both sander and pmemd.
・pH一定シミュレーション用解析ツールの改善
Improved tool for analyzing constant pH simulations
・水素質量の割り当て機能 (ParmEd)
Hydrogen mass repartitioning is supported via ParmEd.
・chamber-styleトポロジーファイルのサポート (ante-MMPBSA.py)
Support for chamber-style topology files with ante-MMPBSA.py
・OpenMMを介してGPUを用いるMD計算 (ParmEd)
ParmEd can run MD calculations on GPUs using OpenMM
・新しい二価金属イオンパラメータや12-6-4 Lennard-Jonesポテンシャルの追加 (sander, pmemd)
New divalent metal ion parameters and a new 12-6-4 Lennard-Jones potential is available for use in sander and pmemd.
・3D-RISMを利用した分子動力学法 (sander)
Molecular Dynamics can be run using 3D-RISM in sander.
・力のトラジェクトリファイル出力への対応
Force trajectories can now be written.
・EMAP、SGLDの実装 (pmemd)
EMAP and SGLD are now implemented in pmemd.
・線形最小二乗法による二面角項のフィッティング (paramfit)
Linear least-squares dihedral term fitting in paramfit.
・NetCDF version 4へ更新
Update bundled NetCDF to NetCDF version 4.
・結晶格子シミュレーションのための新しい解析ツール
New analysis tools for crystal lattice simulations.
・シェル環境設定のための新しい環境リソーススクリプト
New environment resource scripts to help set up shell environment correctly.
・CHARMM力場、およびVMDで生成したトポロジーファイルへの対応強化 (chamber)
Improved support of CHARMM force fields and VMD-generated topology files using chamber.
Amber12の新機能
1. 様々な力場に対応
・電荷固定型のタンパク質の力場であるff12SBが追加されました
・分極ポテンシャルのサポート強化
・脂質の力場Lipid11の他のタンパク質力場との最適化
2. 溶媒和自由エネルギーの計算の精度が向上
・Poisson-Boltzmann方程式に基づいた膜タンパク質、周期境界条件の計算
・強化された3D-RISM積分式モデル(Kovalenko-Hirata理論)
3. サンプリング効率が向上
・Self-guidedランジュバン動力学法(Self-guided Langevin dynamics)
・加速化分子動力学法(Accelerated molecular dynamics)
4. 更新手続きが簡単に
・インストール作業の簡素化、自動更新への対応
5. 量子計算との連携が密に
・d軌道を用いる半経験的量子計算の実装(AM1/dとPM6)
・QM/MM計算での外部量子計算ソフトウエアとの連携
6. CPU版とGPU版両方のpmemdへのsander搭載機能の実装
・温度レプリカ交換法
・等方性周期和
・加速化分子動力学法
・GPU上でNMRoptを用いたさまざまなハーモニックな拘束の対応
7. ハミルトニアンモデルを変更する自由エネルギー計算がより高精度に
・原子の出現・消滅に対応する手法を含む自由エネルギー計算
・レプリカ交換シミュレーションとの連携強化
8. 実験情報を用いたシミュレーション
・電子密度マップ(cryo EM/MT実験など)を束縛条件に指定
・シミュレーション中でグループを固定(もしくは一部を自由に)
Amber11の新機能
2010年4月25日にリリースされたAmber11は、Amber10(2008年4月リリース)から大幅にアップデートされました。以下はAmber.orgに掲載されているAmber11の変更点の和約です。
- CMAP捩れポテンシャルを含む、固定電荷のCHARMM力場のほとんどに対応します。有機分子用の一般的なAmber力場(GAFF)もアップデートしています。
- ペプチドやたんぱく質に最適化された一般化ボルン溶媒モデルのパラメータが利用可能になりました。
- 数値的なポアソン-ボルツマン溶媒の取り扱いに拡張オプションができました。
- 溶媒効果を 3D-RISM 積分方程式モデルを用いて評価できるようになりました。Kovalenko-HirataらのClosure近似が使われています。
- 可視化ソフトとして、Chimera 及び UCSF DOCK との親和性が向上しています。
- ハミルトニアンを変更して、自由エネルギー計算を簡易化する手法が利用可能となりました。
- コンフォメーション遷移に対してエネルギー障壁の低い経路を探索する方法として、Nudged Elastic band 法を新しく実装しました。系の一部にのみ適用することも、explicitな溶媒シミュレーションに用いることもできます。
- pH 一定のシミュレーションと MMPB/SA 自由エネルギー計算のためのスクリプトがアップデートされました。
- 極性のある系についての IPS(Isotropic periodic sum)法と同時に、それと対になる IPS-DFFT 法が実装されました。
- pmemdを用いた MD シミュレーションで NVIDIAのGPU を利用可能となりました。CPU での計算にくらべ、大幅なスピードアップを図ることができます。
Amber10で追加された機能
■力場
- 新しく多数のタイプの力場を追加(水・イオンの新しいモデル、核酸・炭水化物のパラメータをアップデート)
- RenとPonderによるAMOEBA分極ポテンシャルの並列化対応
- 化学反応のための近似ポテンシャルの構築に利用するEmpirical Valence Bond(EVB)モデルの改良
■QM/MMシミュレーション
- 周期的溶媒BOXまたは一般化Born溶媒モデルにおけるDFTB計算が可能に
- コードの高速化、部分的な並列化対応。
■ABMD
- サンプリングと自由エネルギーの収束を加速するため、Adaptively biased シミュレーションが利用可能に
■経路積分分子動力学
- 経路積分分子動力学シミュレーションによって平衡カノニカル分布のサンプリングに原子核の運動のニュートン方程式ではなく量子動力学を用いることが可能に
- 平衡論的、速度論的な同位体効果双方を質量についての熱力学的積分により推算
- 反応速度定数をQuntum Instantonモデルから推算
- 近似量子時間相関関数をRing Polymer MDまたはCentroid MDにより推算
■配座クラスタリング分析ツール
- ptrajにおいて新しい配座クラスタリング分析ツール群が利用可能に
■自由エネルギーツール
- 単一、二重トポロジー双方に利用できるタンパク質ミューテーションの設定が著しく簡略化
- 人為的なダミー原子を必要としないで原子の生成や消滅のある系をサンプリングするためにソフトコアポテンシャルを導入
■レプリカ交換法
- 通常のレプリカ交換法コードの改良
- 非Boltzmann型リザーバをサポート
- Explicitな溶媒中の大規模な系で要求されるレプリカ数を削減するためのハイブリッド溶媒モデル
■pmemd
- pmemdの拡張によるスピード、並列性能の著しい改良
- 一般化Born法への対応
- TIP4PやTIP5Pのような非中心電荷への対応
■LMOD探索
- 低振動数基準モードによるlow-mode(LMOD)配座探索ツールを統合
Amber入門
1. Amberプログラム解説
Amberは生体分子系のシミュレーションを行うための複数のプログラムからなるソフトウェアパッケージです。用途で大別して入力準備プログラム、シミュレーションプログラム、結果解析プログラムから構成されています。それぞれの用途でも複数のプログラムが用意されています。それぞれの概要は以下の通りです。
入力準備プログラム
LEaP :
Amberのための新しい系を作ったり、既存の系を修正したりするための基本プログラムです。
LEaPは以前のバージョンのprep, link, edit, parmの機能を統合したものです。
ANTECHAMBER :
Antechamberファミリーソフトウェアパッケージのメインプログラムです。分子フォーマット変換、原子タイプ指定、電荷生成を自動的に行うため、標準的な核酸やタンパク質を多く含んだ系ならば、LEaPのインプットファイルを準備するのに役立ちます。
シミュレーションプログラム
ANDER :
エネルギー最小化と分子動力学法を行うための基本となるプログラムです。
エネルギー最小化ではエネルギー勾配の平均値が十分低くなるまで、エネルギーが下がる向きに繰り返し原子を動かすことによって構造を緩和します。
分子動力学法ではNewton運動方程式を積分することによって系の配置を生成します。
分子動力学法はエネルギー最小化より多くの配置空間をサンプルします。小さなエネルギー障壁を構造が超えることを可能にします。
配置は後々の解析のために、シミュレーション中に一定間隔で保存されます。熱力学積分を行って基本的な自由エネルギー計算をします。
より精巧な配置空間探索とモデリングの分子動力学法研究もSANDERモジュールを使って実行することができます。
これはいろいろな束縛条件を基本的な力場に加えることを可能にします。特にNMR構造詳細化に関与するタイプの計算のために設計されました。
PMEMD :
並列化効率と速度に関して最適化を図ったバージョンのsanderです。
プログラムの名称は”Particle Mesh Ewald Molecular Dynamics”に基づいており、この種のシミュレーションに限定されています。
入出力ファイルはsanderのものからほんの少しだけしか変更がありません。
NMODE:
準Newton Raphsonによる二次微分を利用したエネルギー最小化と振動解析のプログラムです。
NMODEは、系の多数の熱化学特性とノーマルモードを計算することができます。
他の機能として「 Langevin モード」(連続溶媒への粘性結合を含めたノーマルモード)を計算することが可能であり、エネルギー最小構造と同様に遷移状態を見つけるテクニックを備えます。
結果解析プログラム
PTRAJ :
MDシミュレーションによって(あるいは種々の他のソースから)生成された軌跡(trajectory)や座標を解析したり処理したりする汎用のユーティリティプログラムです。
・重ね合わせ
・座標の抽出
・結合長、結合角、二面角の計算
・原子の座標変位
・相関関数
・水素結合解析
等をの実行します。
同じ実行ファイルが名称をrdparmとして実行されるとき、prmtopファイルを検査したり修正したりすることが出来ます。
MM-PBSA :
連続溶媒モデルを利用した分子動力学シミュレーションからのスナップショットのエネルギー解析を自動化するスクリプトです。
2. Amber利用法概略
新たにAmberを利用されようとするお客様にAmberプログラム利用の流れを説明します(概略の説明ですので個々のプログラムの利用に当たっては個別プログラムのドキュメントを参照してください)。
どういった手順からAmberのシミュレーションを始めるべきかを理解することが第一の問題です。
まず最初にどのシミュレーションプログラム(sander, pmemd, nmode)を利用するかを理解する必要があります。
利用するプログラムが定まりましたら、次に全てのシミュレーションプログラムで必要とする情報を用意します。
全てのシミュレーションプログラムで必要とする情報は以下の通りです。
(1) 系の各原子の三次元座標
X線結晶解析、NMR分光法、モデルビルデングによってProtein Databank (PDB)で作成します。
これらのモデリングタスクの多くに対して、プログラムLEaPは実行プラットホームを供給します。
場合によっては他のプログラムを考慮しなければならないこともあります。
(2) "トポロジー": 結合, 原子名, 原子タイプ, 残基名, 電荷
これらの情報はamber8/dat/leap/prepディレクトリにあるデータベースから得ることが出来ます。
Amber8のマニュアルのChapter 2に詳細な記述があります。
そこには標準アミノ酸、N末端やC末端の荷電アミノ酸、DNA、RNA、通常の糖、等のトポロジーを含んでいます。
データベースはこれらの単量体ユニットのデフォルト内部座標を含んでいます、しかし通常、座標情報はPDBファイルから得られます。
(標準データベースで見いだされない)他の分子のためのトポロジー情報は、通常antechamberを使って利用者が作成し、利用者作成の「残基ファイル」に保存します。
(3) 力場: 系における結合、結合角、二面角、原子タイプなどのパラメータ
いくつかの標準的な力場パラメータはamber8/dat/leap/parmディレクトリに納められています。やはりAmber8のマニュアルのChapter 2に詳細な記述があります。
これらのファイルはタンパク質や核酸についてそのまま利用することもありますし、利用者が標準の力場に対して修正を行ったものを用意して利用することもあります。
(4) コマンド:シミュレーションのオプションと状態変数の指定
これらはmdinと名前を付けたsander, pmemd, nmodeの各プログラムの入力ファイルで指定します。
以上の情報は通常、LEaP、antechamberの入力ファイル作成プログラムを利用して準備します。
準備できましたらシミュレーションプログラム(sander, pmemd, nmode)を実行します。
実行が完了した結果、途中の経過等を解析プログラム(PTRAJ, MM-PBSA)で解析します。
これまでの流れは次の図のようにまとめられます。
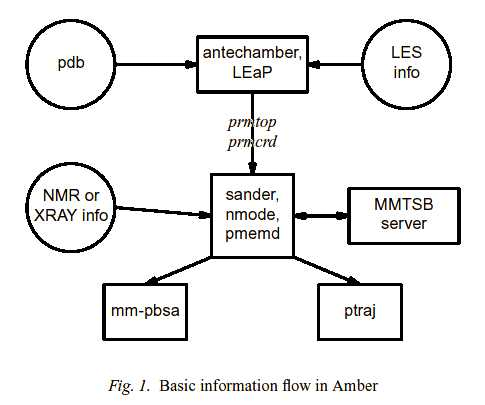
Amber8マニュアルより引用
Amber応用例
- Construction, MD Simulation, and Hydrodynamic Validation of an All-Atom Model of a Monoclonal IgG Antibody
J. Paul Brandt, Thomas W. Patapoff, and Sergio R. Aragon Biophys J. 2010 August 4 99(3):905-913.
Amber検証日記
2011年7月14日
Amber11をambertools ver1.5を使用するようにアップデートしようとお考えの方も多い事かと思います。弊社もそうしたご要望に対応するべく検証作業を行ってきたのですが、なかなか今すぐシステム構築対応は出来ませんでした。出来なかったのには理由があります。
それはamber11をビルドして、まずはとシリアル版でテストを行った所、エラーしてしまうテストがあったのです。
具体的にはsander_pbsa_frc以下のものなのですが、これがどうしても上手くゆきません。
amberのメーリングリストでも多数の悲鳴のようなヘルプを求める内容が散見されていて、シリアル版のテストでエラーが出てしまうのは弊社の技術の問題ではなく、新しいambertools 1.5で何かが起きているのではないかと思われました。
どうしても、新規に作成されたプログラムには、エラーが含まれてしまう事が多いので、ambertoolsを開発しているチームも例外では無かったという事だと思います。特に、ambertoolsはGPLになりましたから、開発を以前のように閉じた環境で行っている訳ではないので、やむを得ないでしょう。
しかし、amberの開発チームは、丁寧なパッチリリースを行いますから、これはパッチのリリースを待とうという事にしました。
信じて待った甲斐もあり、ambertools 1.5のpatch6でこの問題に対応したパッチをリリースしたとのアナウンスがあり、やっとこれで、ambertools 1.5を含むシステム構築が可能になると喜んでパッチをあててテストを行いました。
結果はまったく変わりません。
それではpatchは何を?
この対応パッチは以下の内容をビルドする時に出力するものでした。
NOTE: Because PBSA has changed since Amber 11 was released, some
tests are known to fail and others are known to quit in error.
These can be safely ignored.
Tests that error: Tests in $AMBERHOME/test/sander_pbsa_frc
Run.argasp.min Run.dadt.min Run.dgdc.min
Run.lysasp.min Run.polyALA.min Run.polyAT.min
Run.argasp.min Run.dadt.min Run.dgdc.min
Run.lysasp.min Run.polyALA.min Run.polyAT.min
Run.argasp.min Run.dadt.min Run.dgdc.min
Run.lysasp.min Run.polyALA.min Run.polyAT.min
Tests that produce possible FAILUREs:
cd sander_pbsa_ipb2 && ./Run.110D.min
cd sander_pbsa_lpb && ./Run.lsolver.min (only some of them fail here)
cd sander_pbsa_tsr && ./Run.tsrb.min
cd sander_pbsa_decres && ./Run.pbsa_decres
mm_pbsa.pl tests 02, 03, and 05
つまり、エラーが出るのは仕様のようです。
実はambertoolsがこうしたamber本体と食い違う事は以前、amber10のある時期にもありました。
その時期とは開発チームがamber11の開発を精力的に行っていた時期です。
この時は、amber11の為のambertoolsのプレリリースに近い扱いだったのですが、今回もそうした思惑がamberの開発チームにはあるのかもしれません。
ambertools ver1.4でテストでエラーの出ない形でシステム構築は当然、可能ですから、もし、こちらがご希望の場合は、弊社営業にご相談下さい。
2011年2月24日
ambertools 1.4のpatch 18がリリースされました。sleapに幾つかコマンドが追加されました。
2011年2月3日
2011年2月3日現在、Amber11の最新パッチはPatch 12です。Amber11のpatchでは大きく分けて、sander等の機能やバグフィックスに関するパッチと、GPU化に関係するパッチに分かれま す。patch 3, 4, 9, 11, 12はcuda対応でのパッチです。また、パッチを当てるツール、apply_bugfix.xは、こっそりと01/07/2011の日付けで、アップグ レードされています。
また、ambertools 1.4用に、gauusian 09 B.01に関係するツール、fixreadinesp.shが提供されています。
2010年7月28日
Amber11 Patch6 がでています。sander.PUPILに関するものだったため、改めて
PUPILの準備。PUPILがJAVAを使うため、必要な環境整備や設定、PUPIL v1.3.0
のビルドに成功。しかし、肝心のsander.PUPILがビルドできない。Try&Error
を繰り返したものの、どうも修正Patchに問題がありそうな予感がします。



お問い合わせ
平日9:30~17:30 (土曜日、日曜日、祝祭日、年末年始、夏期休暇は、休日とさせていただきます。)